محلل القيم الذاتية الكمومي التباديلي (VQE)
لهذه الوحدة، يجب أن يمتلك الطلاب بيئة Python تعمل بشكل صحيح، مع تثبيت أحدث إصدارات الحزم التالية:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
لإعداد هذه الحزم وتثبيتها، راجع دليل تثبيت Qiskit. لتشغيل المهام على حواسيب كمومية حقيقية، يحتاج الطلاب إلى إنشاء حساب IBM Cloud، باتباع الخطوات الواردة في دليل إعداد حساب IBM Cloud الخاص بك.
تم اختبار هذه الوحدة واستخدمت ما يقارب 8 دقائق من وقت وحدة معالجة الكم (QPU). هذا تقدير، وقد يختلف استخدامك الفعلي.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
مقدمة
منذ تطوير النموذج الميكانيكي الكمومي في مطلع القرن العشرين، أدرك العلماء أن الإلكترونات لا تسلك مسارات ثابتة حول نواة الذرة، بل توجد في مناطق احتمالية تُسمى المدارات. تقابل هذه المدارات مستويات طاقة محددة ومنفصلة يمكن للإلكترونات أن تشغلها. تستقر الإلكترونات بطبيعتها في أدنى مستويات الطاقة المتاحة، المعروفة بالحالة الأساسية. غير أنه إذا امتصّ إلكترون طاقةً كافية، يمكنه القفز إلى مستوى طاقة أعلى، فيدخل في حالة مثارة. هذه الحالة المثارة مؤقتة، وسيعود الإلكترون في نهاية المطاف إلى مستوى طاقة أدنى، محرِّرًا الطاقة الممتصة، في الغالب على شكل ضوء. هذه العملية الأساسية لامتصاص الطاقة وانبعاثها مهمة لفهم كيفية تفاعل الذرات وتكوين الروابط.
حين تتحد الذرات لتكوين جزيئات، تتحد مداراتها الذرية لتكوين مدارات جزيئية. يحدد ترتيب الإلكترونات ومستويات طاقتها داخل هذه المدارات الجزيئية خصائص الجزيء الناتج وقوة الروابط الكيميائية. على سبيل المثال، في تكوين جزيء الهيدروجين () من ذرتَي هيدروجين منفردتين، يشغل إلكترون كل ذرة مداراتها الذرية. وعندما تقترب الذرتان من بعضهما، تتداخل هذه المدارات الذرية وتتحد لتكوين مدارات جزيئية جديدة — واحدة ذات طاقة أدنى (مدار رابط) وأخرى ذات طاقة أعلى (مدار مضاد للرابط). سيشغل الإلكترونان، كلٌّ منهما من ذرة هيدروجين، مدار الرابط ذا الطاقة الأدنى تفضيلًا، مما يؤدي إلى تكوين رابطة تساهمية مستقرة تُمسك جزيء معًا. يحدد فرق الطاقة بين الذرات المنفصلة والجزيء المتكوّن، ولا سيما طاقة الإلكترونات في المدارات الجزيئية، استقرار الرابطة وخصائصها.
في الأقسام التالية، سنستكشف هذه العملية لتكوين الجزيئات، مع التركيز على جزيء . سنستخدم حاسوبًا كموميًا حقيقيًا، مقترنًا بتقنيات تحسين كلاسيكية، لإيجاد طاقة هذه العملية البسيطة والجوهرية. ستوفر هذه التجربة عرضًا عمليًا لكيفية تطبيق الحوسبة الكمومية لحل مسائل في الكيمياء الحاسوبية، مع تسليط الضوء على دور طاقة الإلكترونات.
VQE - خوارزمية كمومية تباديلية لمسائل القيم الذاتية
تقنيات التقريب في الكيمياء - المبدأ التباديلي ومجموعة الأساس
لا تقتصر إسهامات إيرفين شرودنغر في ميكانيكا الكم على تقديم نموذج إلكتروني جديد؛ فقد أرسى في جوهره ميكانيكا الموجات بتطويره معادلة شرودنغر الشهيرة المعتمدة على الزمن:
هنا، هو الهاملتوني الذي يمثل الطاقة الكلية للنظام، و هي دالة الموجة التي تحتوي على جميع المعلومات المتعلقة بالحالة الكمومية للنظام. (ملاحظة: هو المشتق الكلي بالنسبة للزمن، ولا نُدرج هنا بشكل صريح قيمة القيمة الذاتية للطاقة .)
ومع ذلك، في كثير من التطبيقات العملية — كتحديد مستويات الطاقة المسموح بها للذرات والجزيئات — نلجأ بدلًا من ذلك إلى معادلة شرودنغر المستقلة عن الزمن (معادلة القيمة الذاتية للطاقة)، التي تُشتق من الصيغة المعتمدة على الزمن بافتراض حالة ثابتة. الحالة الثابتة هي حالة كمومية لا تتغير فيها كثافة الاحتمال لإيجاد جسيم عند نقطة معينة في الفضاء بمرور الوقت.
في هذه الصيغة، تمثل القيمة الذاتية للطاقة المقابلة للحالة الكمومية . يشمل الهاملتوني مساهمات طاقة متنوعة، كالطاقة الحركية للإلكترونات والنوى، والقوى الجاذبة بين الإلكترونات والنوى، والقوى التنافرية بين الإلكترونات.
يتيح لنا حل معادلة القيمة الذاتية للطاقة حساب مستويات الطاقة المكمّمة للأنظمة الذرية والجزيئية. غير أنه بالنسبة للجزيئات، يصعب حلها بشكل دقيق لأن دالة الموجة ، التي تصف التوزيع المكاني للإلكترونات، معقدة وعالية الأبعاد.
لذا يلجأ العلماء إلى تقنيات التقريب للحصول على حلول عملية ودقيقة. في هذا العمل، سنركز على طريقتين رئيسيتين:
-
المبدأ التباديلي
تُقرّب هذه الطريقة دالة الموجة وتضبطها للاقتراب قدر الإمكان من طاقة الهدف، وعادةً ما تكون طاقة الحالة الأساسية للنظام. الفكرة الرئيسية وراء المبدأ التباديلي بسيطة:
- إذا خمّنا دالة موجة ("دالة تجريبية")، فإن الطاقة المحسوبة منها ستكون دائمًا مساوية لطاقة الحالة الأساسية () للنظام أو أعلى منها.
- بضبط المعاملات في الدالة التجريبية، ، يمكننا الحصول على تقريب أفضل وأفضل لطاقة الحالة الأساسية.
- تعتمد دقة هذه الطريقة بشكل كبير على اختيار دالة الموجة التجريبية . فقد تؤدي الدالة التجريبية المختارة بشكل سيئ إلى تقدير للطاقة يبعد كثيرًا عن الدقة.
-
تقريب مجموعة الأساس
تأتي طريقة التقريب الثانية في مرحلة بناء دالة الموجة — مقاربة مجموعة الأساس. في الكيمياء الكمومية، يكاد يكون من المستحيل حل معادلة شرودنغر بدقة تامة للجزيئات. عوضًا عن ذلك، نُقرّب دالة الموجة المعقدة متعددة الإلكترونات ببنائها من دوال رياضية أبسط ومحددة مسبقًا. مجموعة الأساس هي في الأساس مجموعة من هذه الدوال الرياضية المعروفة، التي تتمركز عادةً حول ذرات الجزيء، وتُستخدم كلبنات بناء لتمثيل شكل الإلكترونات وسلوكها في النظام. فكّر في الأمر كمحاولة إعادة خلق تمثال دقيق باستخدام مجموعة من قطع LEGO المعيارية فحسب — كلما زاد عدد أنواع القطع وأحجامها (كلما كانت مجموعة الأساس أكبر)، كان بإمكانك تقريب الشكل الأصلي بدقة أكبر.
غالبًا ما تستلهم دوال الأساس هذه من الحلول التحليلية لأنظمة بسيطة كذرة الهيدروجين، مُتخذةً أشكالًا كالدوال الغاوسية أو دوال نوع سليتر، وإن كانت لا تزال تقريبات. بدلًا من العمل مع المدارات الجزيئية الكاملة "الدقيقة" نظريًا ولكن غير القابلة للمعالجة، نعبّر عنها كتركيبة خطية (مجموع بمعاملات) لهذه الدوال الأساسية. تُعرف هذه الطريقة بمقاربة التركيبة الخطية للمدارات الذرية (LCAO) حين تشبه دوال الأساس المدارات الذرية. بتحسين المعاملات في هذه التركيبة الخطية، يمكننا إيجاد أفضل دالة موجة تقريبية ممكنة وطاقتها ضمن حدود مجموعة الأساس المختارة.
- كلما زاد عدد الدوال المُدرجة في مجموعة الأساس، كان التقريب أفضل، لكن يأتي ذلك على حساب جهد حوسبي أعلى.
- تُعطي مجموعة الأساس الصغيرة تقديرًا تقريبيًا، في حين تُعطي مجموعة الأساس الكبيرة نتائج أكثر دقة على حساب الحاجة إلى موارد حوسبية أكبر.
خلاصة القول، لجعل الحسابات قابلة للتنفيذ وتقليل التكلفة الحوسبية، نستخدم المبدأ التباديلي بتقريب دالة الموجة، مما يُقلل من تعقيد الحوسبة ويسمح بالتحسين التكراري لتقليل الطاقة. في الوقت ذاته، يُبسّط نهج مجموعة الأساس الحسابات بتمثيل المدارات الذرية كتركيبة من دوال محددة مسبقًا، بدلًا من حل دالة موجة مستمرة مباشرةً.
تحقق من فهمك
لنأخذ دالة الموجة التجريبية حيث ثابت تطبيع و معامل قابل للضبط.
(a) طبّع دالة الموجة التجريبية بتحديد بحيث يكون .
الإجابة
لتطبيع دالة الموجة التجريبية المعطاة:
استخدم التكامل الغاوسي:
نضع ثم نحصل على:
(b) احسب القيمة المتوقعة للهاملتوني المعطى بـ حيث ، وهو ما يقابل كمون المذبذب التوافقي البسيط.
الإجابة
هاملتوني المذبذب التوافقي هو:
القيمة المتوقعة للطاقة الحركية
بأخذ المشتق الثاني:
وبذلك:
باستخدام نتائج التكامل الغاوسي القياسية:
القيمة المتوقعة لطاقة الوضع
باستخدام:
نحصل على:
القيمة المتوقعة للطاقة الكلية
(c) استخدم المبدأ التباديلي لإيجاد القيمة المثلى لـ بتقليل .
الإجابة
تحسين للحصول على الطاقة الدنيا
اشتقاق
بالحل:
بالتعويض بـ في :
وهو ما يتوافق مع طاقة الحالة الأساسية الدقيقة للمذبذب التوافقي الكمومي.
VQE (Variational Quantum Eigensolver)
محلل القيم الذاتية الكمومي التباديلي (VQE) هو الأسلوب الرئيسي الذي سنستخدمه لاستكشاف عملية ، وهنا سنلقي نظرة على ما هو VQE وكيف يعمل. لكن لنتوقف أولاً ونتأمل شيئاً بالغ الأهمية من خلال سؤال المراجعة التالي.
تحقق من فهمك
إذا كانت لدينا بالفعل هذا العدد الكبير من الاستراتيجيات لمعالجة مسائل الكيمياء، فلماذا نحتاج إلى حاسوب كمومي؟ وما الغرض من استخدام الحاسوب الكمومي والحاسوب الكلاسيكي معاً؟
الإجابة
تمتلك الحوسبة الكمومية فرصة لإحداث ثورة في الكيمياء من خلال معالجة المسائل التي تعجز عنها الحواسيب الكلاسيكية بسبب التوسع الأسي للحالات الكمومية. أشار ريتشارد فاينمان بشكل مشهور إلى أنه لمحاكاة الطبيعة، يجب أن تكون الحسابات كمومية أيضاً [المرجع 1].
على سبيل المثال، تحتاج محاكاة الكافيين باستخدام أبسط مجموعة أساسية (STO-3G) إلى بت، وهو عدد أكبر بكثير من إجمالي عدد النجوم في الكون المرئي () [المرجع 2]. يستطيع الحاسوب الكمومي وصف المدارات الإلكترونية للكافيين باستخدام 160 كيوبت.
تعالج الحواسيب الكمومية بطبيعتها التفاعلات الكمومية باستخدام التراكب والتشابك، وهو ما يوفر طريقة واعدة لتمكين محاكاة جزيئية دقيقة. علاوة على ذلك، يمكننا الجمع بين مزايا الحواسيب الكمومية (محاكاة الإلكترونات) والحواسيب الكلاسيكية (المعالجة المسبقة/اللاحقة للبيانات، وإدارة عملية الخوارزمية، والتحسين، وما إلى ذلك). ومن المتوقع أن تُعزز هذه الأساليب اكتشاف المواد، وتصميم الأدوية، والتنبؤ بالتفاعلات، مما يُقلل من التجارب المكلفة القائمة على التخمين. [المرجع 3][المرجع 4]
إذا أردت معرفة سبب الحاجة إلى الحواسيب الكمومية لمعالجة مسائل الكيمياء، ولماذا نستخدم موارد الحوسبة الكمومية والكلاسيكية معاً، اطلع على المقالات التالية:
لنعد الآن إلى VQE.
يجمع VQE بين قوة الحواسيب الكمومية والكلاسيكية، معتمداً بشكل أساسي على المبادئ التباديلية للحصول على طاقة الحالة الأساسية للنظام. لفهم VQE، قسّمه أولاً إلى ثلاثة أجزاء:
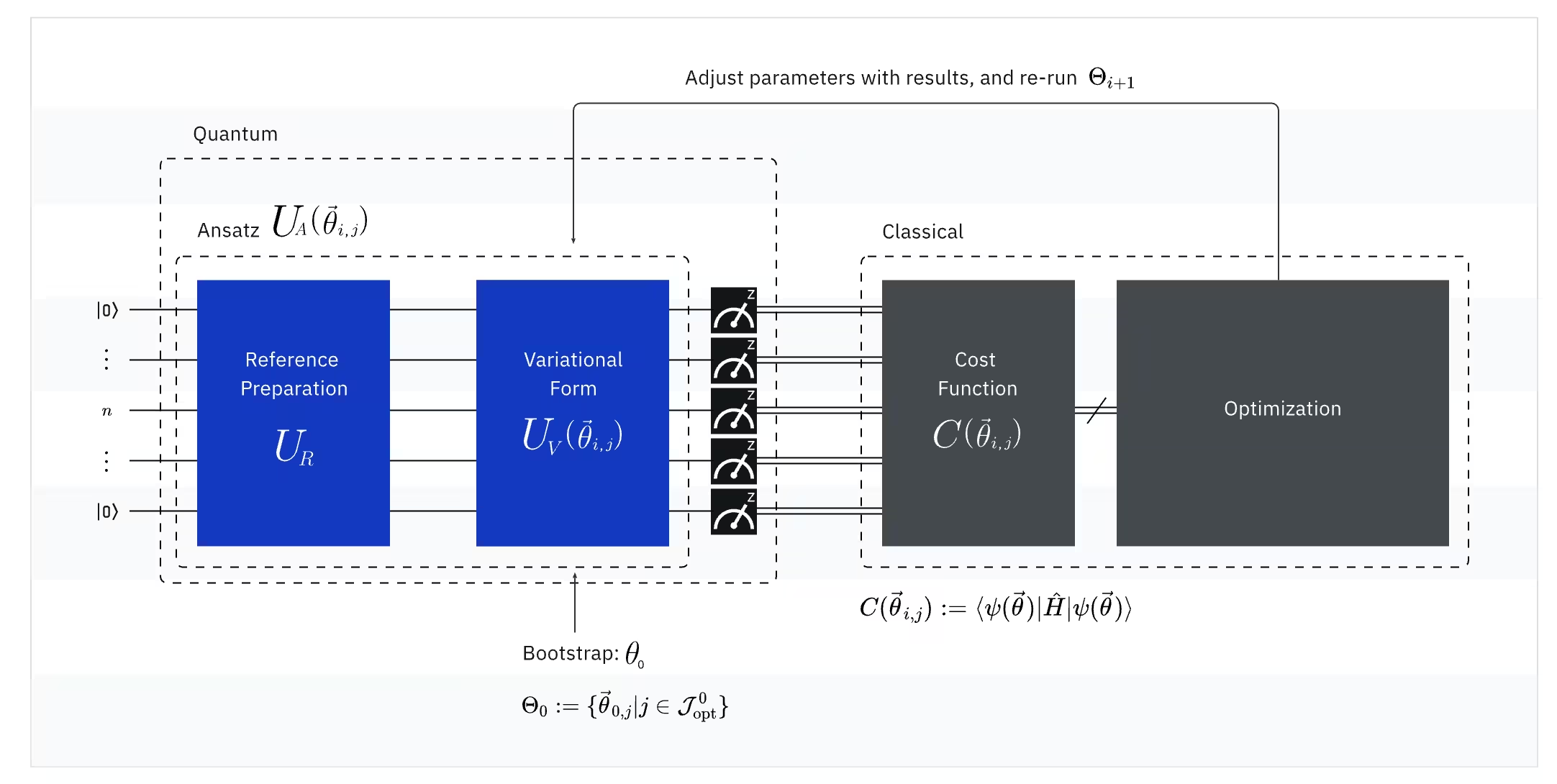
(كمي) المؤثر الرصدي: هاملتوني الجزيء (طاقة الجزيء)
في VQE، يُعدّ هاملتوني الجزيء/الذرة مؤثراً رصدياً، أي أننا نستطيع قياس قيمته من خلال تجربة. هدفنا هو إيجاد أدنى طاقة ممكنة (طاقة الحالة الأساسية) للجزيء. لتحقيق ذلك، نستخدم حالة كمومية تجريبية، يُولّدها دائرة كمومية ذات معاملات قابلة للضبط (ansatz). نقيس المؤثر الرصدي ونحسّن الحالة الكمومية حتى نصل إلى أدنى طاقة ممكنة.
تحدد مجموعة الأساس المستخدمة لهاملتوني الجزيء عدد الكيوبتات المطلوبة وتؤثر مباشرة على دقة VQE. يُعدّ اختيار مجموعة الأساس المناسبة أمراً بالغ الأهمية لتحقيق التوازن بين الكفاءة والدقة. ولتبسيط الحسابات دون تغيير مجموعة الأساس، يمكننا استخدام استراتيجيات مثل فرض التماثل وتقليل الفضاء الفعّال. كثير من الجزيئات لها أشكال متماثلة (كالفراشة أو ندفة الثلج)، مما يعني أن بعض أجزائها تتصرف بالطريقة ذاتها. بدلاً من حساب كل شيء بشكل منفصل، يمكننا التركيز فقط على الأجزاء الفريدة، مما يوفر الموارد الكمومية، وبالتالي الاستفادة من التماثل. في تقليل الفضاء الفعّال، نأخذ في الاعتبار المدارات المهمة فقط، إذ لا تؤثر جميع الإلكترونات بشكل ملحوظ على طاقة الجزيء. تظل الإلكترونات القريبة من النواة غير متغيرة إلى حد بعيد، في حين تؤثر إلكترونات أخرى على الروابط. بتطبيق هذه الأساليب، يمكننا جعل VQE أكثر كفاءة مع الحفاظ على الدقة.
بعد الحصول على هاملتوني الجزيء باستخدام مجموعة الأساس المناسبة والاستراتيجيات المذكورة أعلاه، نحتاج إلى تحويل هذا الهاملتوني إلى صيغة مناسبة للحواسيب الكمومية. قد يكون رسم خرائط المسائل إلى مؤثرات باولي أمراً بالغ التعقيد. ينطبق هذا بشكل خاص على الكيمياء الكمومية التي تتعامل مع جسيمات غير قابلة للتمييز (إلكترونات)، نظراً لأن الكيوبتات قابلة للتمييز. لن نخوض في تفاصيل رسم الخرائط هنا، لكننا نحيلك إلى الموارد التالية. يمكن الاطلاع على مناقشة عامة لرسم خريطة المسألة إلى مؤثرات كمومية في الحوسبة الكمومية التطبيقية. كما يمكن العثور على مناقشة أكثر تفصيلاً حول رسم خرائط مسائل الكيمياء إلى مؤثرات كمومية في الكيمياء الكمومية مع VQE.
في هذه الوحدة، سنزودك بهاملتونيات (أحادية الكيوبت) مناسبة لـ و حتى نتمكن من التركيز على استخدام الحاسوب الكمومي. تم إعداد هذه الهاملتونيات أحادية الكيوبت باستخدام مجموعة الأساس STO-6G ورسم خريطة جوردان-فيغنر، وهو أبسط رسم خريطة بالتفسير الفيزيائي الأيسر، إذ يربط إشغال مدار دوراني واحد بإشغال كيوبت واحد. كما استخدمنا تقنية تقليل الكيوبتات بالاستفادة من تماثل الهاملتوني، التي تستخدم الأنماط في سلوك إشغالات الدوران لتقليل عدد الكيوبتات. بالنسبة لجزيء ، نفترض أن المسافة بين ذرتي الهيدروجين هي 0.735 .
(كمومي) الـ Ansatz: دالة الموجة التجريبية (كيفية بناء حالة كمومية بسيطة باستخدام دائرة كمومية)
في VQE، يتكون الـ ansatz (الجمع: ansätze) من عنصرين رئيسيين. الأول هو تحضير الحالة الابتدائية، الذي يُهيئ حالة الكيوبت من خلال تطبيق بوابات كمومية دون معاملات تباديلية. أما العنصر الثاني فهو الدائرة الكمومية ذات المعاملات، وهي دائرة كمومية خاصة بمعاملات قابلة للتعديل، تشبه مقابض الضبط في جهاز راديو. ستُستخدم هذه المعاملات في الجزء الأخير — المحسّن الكلاسيكي — لمساعدتنا على الوصول إلى أفضل حالة أساسية ممكنة.
في قسم المبدأ التباديلي، تعلمنا أن جودة الحالة التجريبية تؤثر على جودة نتائج الخوارزمية التباديلية. هذا يعني أن اختيار ansatz جيد أمر مهم في VQE. مرة أخرى، هذا موضوع غني ومعقد. لن نغطي الأنواع المختلفة من ansatz أو أصولها هنا. إذا كنت مهتماً بمعرفة المزيد عن الدوائر الكمومية ذات المعاملات والـ ansatz، يمكنك استكشاف درس Ansatz والصيغة التباديلية من دورة تصميم الخوارزميات التباديلية، الذي يقدم شروحاً وأمثلة تفصيلية لأنواع ansätze المختلفة.
بما أننا سنستخدم هاملتونياً أحادي الكيوبت في هذه الوحدة، نحتاج إلى دائرة كمومية ذات معاملات أحادية الكيوبت كـ ansatz. سنرى ثلاثة أنواع من ansätze أحادية الكيوبت في القسم التالي. سنقارنها ونناقش الاعتبارات الرئيسية في اختيار الـ ansatz.
(كلاسيكي) المحسّن: الضبط الدقيق للدائرة الكمومية
بمجرد أن يقيس الحاسوب الكمومي طاقة المؤثر الرصدي من الـ ansatz، تُرسل معاملات الـ ansatz وقيمة الطاقة إلى المحسّن الكلاسيكي للضبط. تُنفَّذ عملية التحسين هذه على حاسوب كلاسيكي، باستخدام حزم علمية عامة الغرض مثل SciPy عادةً.
يتعامل المحسّن الكلاسيكي مع الطاقة المقاسة باعتبارها دالة تكلفة. في مسائل التحسين، دالة التكلفة (التي تُسمى أحياناً دالة الهدف) هي دالة رياضية تقيس مدى "جودة" حل معين. هدف المحسّن هو إيجاد مجموعة المعاملات التي تُقلل هذه الدالة إلى الحد الأدنى. في سياق إيجاد طاقة الحالة الأساسية لجزيء ما، تعمل الطاقة ذاتها كدالة تكلفة — نريد إيجاد المعاملات لدائرتنا الكمومية ("حلنا") التي تُعطي أدنى طاقة ممكنة. يستخدم المحسّن الكلاسيكي قيمة الطاقة المقاسة (التكلفة) ويحدد المجموعة التالية من المعاملات المحسّنة لـ ansatz الكمومي. تُرسل هذه المعاملات المحدّثة إلى الدائرة الكمومية، وتتكرر العملية. مع كل تكرار، يضبط المحسّن الكلاسيكي المعاملات في محاولة لخفض الطاقة (تصغير دالة التكلفة) حتى يتحقق معيار تقارب محدد مسبقاً، مما يضمن من الناحية المثالية إيجاد أدنى طاقة ممكنة (المقابلة للحالة الأساسية للجزيء عند مسافة الرابطة ومجموعة الأساس تلك).
ثمة كثير من استراتيجيات التحسين التي توفرها الحزم العلمية مثل SciPy. يمكنك الاطلاع على المزيد في درس حلقات التحسين من دورة تصميم الخوارزميات التباديلية. سنستخدم هنا COBYLA (التحسين المقيّد بالتقريبات الخطية)، وهو خوارزمية تحسين مناسبة لمشاهد الطاقة المعقدة. على وجه الخصوص، لا تحاول COBYLA حساب التدرج للدالة قيد الدراسة؛ لذا تُسمى محسّناً خالياً من التدرج. تخيل أنك تحاول إيجاد أعلى قمة في سلسلة جبال وعيناك مغمضتان. بما أنك لا ترى المشهد كاملاً، تخطو خطوات صغيرة في اتجاهات مختلفة، مع التحقق مما إذا كنت تصعد أم تنزل. تعمل COBYLA بطريقة مماثلة — تتنقل عبر فضاء المعاملات، تختبر قيماً مختلفة، وتُحسّن النتيجة تدريجياً حتى تجد الأفضل.
أنت الآن مستعد لإجراء حساب VQE. لذا، جرّب سؤال المراجعة أدناه، الذي يستعرض العملية الكاملة.
تحقق من فهمك
أكمل الفراغات بالمصطلحات الصحيحة لإتمام ملخص عملية VQE، ثم انقر للتحقق من إجاباتك.
VQE هو خوارزمية كمومية تباديلية، تجمع بين قوة (1) ________ والحوسبة الكلاسيكية، وتُستخدم لإيجاد (2) __________ للجزيء. تبدأ العملية بتعريف (3) __________، الذي يمثل الطاقة الكلية للنظام ويعمل كمؤثر رصدي في القياسات الكمومية. بعد ذلك، نُعدّ (4) __________، وهو دائرة كمومية ذات معاملات قابلة للتعديل تمثل دالة الموجة التجريبية للجزيء. يتم تحسين هذه المعاملات باستخدام (5) __________، وهو خوارزمية كلاسيكية تضبط المعاملات بشكل تكراري لتصغير الطاقة المقاسة. في المناقشة أعلاه استخدمنا محسّن (6) __________، الذي يُنقّح معاملات الـ ansatz دون الحاجة إلى حسابات المشتقات. تستمر العملية حتى نصل إلى (7) __________، أي إيجاد أدنى طاقة ممكنة للجزيء.
بنك الكلمات:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
الإجابة
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
حساب طاقة الحالة الأساسية لذرة الهيدروجين باستخدام VQE
لنستخدم الآن ما تعلمناه لحساب طاقة الحالة الأساسية لذرة الهيدروجين. طوال الوحدة، سنستخدم إطاراً للحوسبة الكمومية يُعرف بـ "أنماط Qiskit"، الذي يُقسّم سير العمل إلى الخطوات التالية:
- الخطوة 1: رسم خريطة المدخلات الكلاسيكية إلى مسألة كمومية
- الخطوة 2: تحسين المسألة للتنفيذ الكمومي
- الخطوة 3: التنفيذ باستخدام أوليات Qiskit Runtime
- الخطوة 4: المعالجة اللاحقة والتحليل الكلاسيكي

سنتبع هذه الخطوات بشكل عام.
لنبدأ بتحميل بعض الحزم الضرورية، بما فيها أوليات Qiskit Runtime. سنختار أيضاً الحاسوب الكمومي الأقل ازدحاماً المتاح لنا.
يوجد أدناه كود لحفظ بيانات اعتمادك عند الاستخدام الأول. احرص على حذف هذه المعلومات من الدفتر بعد حفظها في بيئتك، حتى لا تُشارَك بيانات اعتمادك بطريق الخطأ عند مشاركة الدفتر. راجع إعداد حساب IBM Cloud وتهيئة الخدمة في بيئة غير موثوقة للمزيد من التوجيهات.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
ستتيح لك الخلية أدناه التبديل بين استخدام المحاكي أو الأجهزة الحقيقية طوال الدفتر. نوصي بتشغيلها الآن:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
الخطوة 1: رسم خريطة المسألة إلى دوائر كمومية ومؤثرات
نبدأ حساب VQE بتعريف هاملتوني جزيء الهيدروجين () عند مسافة رابطة محددة. يمثل هذا الهاملتوني الطاقة الكلية للنظام من حيث مؤثرات الكيوبت، وقد تم اشتقاقه ورسم خريطته من النظام الجزيئي باستخدام إجراء قياسي: 1) توظيف مجموعة الأساس STO-6G (مجموعة محددة من الدوال الرياضية تُستخدم لتقريب المدارات الإلكترونية)، 2) تطبيق رسم خريطة جوردان-فيغنر (تقنية لترجمة مؤثرات الفرميون التي تصف الإلكترونات إلى مؤثرات كيوبت)، و3) إجراء تقليل الكيوبتات باستخدام التكافؤ (parity) للهاملتوني لتبسيط المسألة.
كما أوضحنا سابقاً، تعتمد طاقات الحالة الأساسية المحسوبة اعتماداً كبيراً على اختيار مجموعة الأساس والهندسة الجزيئية (كمسافة الرابطة). لهذا التكوين المحدد وبعد هذه التحويلات، يكون هاملتوني الكيوبت الناتج بسيطاً:
هنا، يمثل مؤثر الهوية و يمثل مؤثر باولي-Z، يعملان على كيوبت واحد. المعاملات مشتقة من التكاملات المحسوبة باستخدام مجموعة الأساس STO-6G عند مسافة الرابطة هذه مع التحويل المناسب.
مع تحديد هذا الهاملتوني، يمكننا الآن استخدام VQE لحساب طاقة حالته الأساسية. من المفيد مقارنة طاقة الحالة الأساسية المحسوبة بالقيم المتوقعة. لذرة هيدروجين واحدة ومعزولة (H)، تكون طاقة الحالة الأساسية بالضبط -0.5 هارتري (في غياب التأثيرات النسبية). لنحسب طاقة الحالة الأساسية الدقيقة لهاملتوني الكيوبت المحدد كما هو معرّف أعلاه، ونقارنها بالقيم المعروفة ذات الصلة.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
بعد ذلك، نحتاج إلى دائرة كمومية ذات معاملات، أي ansatz، لتحضير دالة موجة تجريبية للحالة الأساسية. الهدف هو إيجاد المعاملات التي تُقلل قيمة توقع الطاقة . يُعدّ اختيار الـ ansatz أمراً بالغ الأهمية لأنه يحدد مجموعة الحالات الكمومية الممكنة التي يمكن لدائرتنا إعدادها. الـ ansatz "الجيد" هو الذي يكون مرناً بما يكفي لتمثيل حالة قريبة جداً من الحالة الأساسية الحقيقية للهاملتوني الذي ندرسه، لكنه ليس معقداً لدرجة تستلزم معاملات كثيرة أو دائرة عميقة جداً للحواسيب الكمومية الحالية.
هنا، سنجرب ثلاثة ansätze مختلفة أحادية الكيوبت لنرى أيها يوفر "تغطية" أفضل للحالات الكمومية الممكنة لكيوبت واحد. تشير "التغطية" إلى نطاق الحالات الكمومية التي يمكن لدائرة الـ ansatz إنتاجها بتغيير معاملاتها.
سنستخدم ثلاثة ansätze تعتمد على تركيبات مختلفة من بوابات الدوران أحادية الكيوبت:
- ansatz بوابة دوران حول محور واحد: يستخدم هذا الـ ansatz دورانات حول محور واحد فقط (). على كرة بلوخ، يقابل هذا التحرك على طول دائرة محددة فقط. وهو الأقل مرونة ويغطي مجموعة محدودة من الحالات.
- ansätze بوابتي دوران حول محورين: تجمع هذه الـ ansätze بين الدورانات حول محورين مختلفين ( و ). هذا يتيح لنا الوصول إلى جزء أكبر من كرة بلوخ مقارنة بالدوران أحادي المحور.
من خلال مقارنة نتائج VQE المحصّلة بهذه الـ ansätze الثلاثة، يمكننا رؤية كيف تؤثر مرونة الـ ansatz وتغطيته لفضاء الحالات على قدرتنا على إيجاد طاقة الحالة الأساسية الحقيقية لهاملتونينا المبسّط. الـ ansatz الأكثر مرونة لديه إمكانية إيجاد تقريب أفضل، لكنه قد يكون أيضاً أصعب على المحسّن الكلاسيكي.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
لنولّد الآن 5000 رقم عشوائي لكل معامل ونرسم توزيع الحالات الكمومية العشوائية التي ولّدتها الـ ansätze الثلاثة بهذه المعاملات العشوائية. يمكنك التفكير في هذه المعاملات كدورانات حول محاور مختلفة على سطح كروي. لرؤية توزيع الحالة الكمومية، سنستخدم كرة بلوخ، وهي كرة ثلاثية الأبعاد تُظهر حالة كيوبت واحد. أي نقطة على الكرة تمثل حالة ممكنة للكيوبت، حيث يشبه القطبان الشمالي والجنوبي الحالتين الكلاسيكيتين "0" و"1"، لكن الكيوبت يمكن أن يكون في أي مكان بينهما، مما يُظهر خصائص كمومية خاصة كالتراكب. أولاً، أعدّ الدوال اللازمة لرسم كرة بلوخ ثلاثية الأبعاد وجهّز 5000 معامل عشوائي.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
لنرَ كيف يعمل أول ansatz لدينا.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
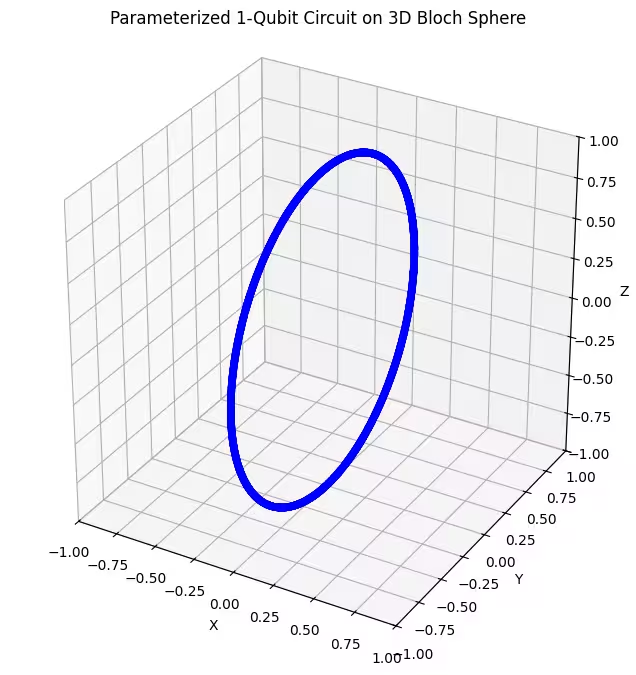
نلاحظ أن أول ansatz يُعيد حالات كمومية موزعة على شكل حلقة على كرة بلوخ. هذا منطقي، لأننا أعطينا الـ ansatz معاملاً دورانياً واحداً فقط. لذا يستطيع إنتاج حالات مُدارة حول محور واحد فقط. الانطلاق من النقطة والدوران حول محور واحد سيُنتج دائماً حلقة. لنتحقق الآن من ansatz الثاني، الذي يضم بوابتي دوران متعامدتين - Rx و Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
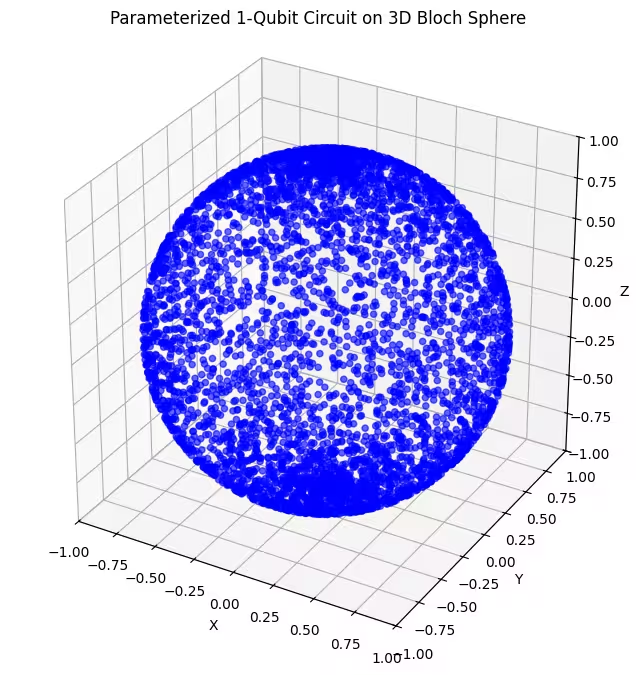
هنا، نرى أن ansatz الثاني يغطي جزءاً أكبر من كرة بلوخ - لكن لاحظ أن النقاط أكثر تركزاً عند القطبين وأكثر انتشاراً عند خط الاستواء. حان الوقت الآن للتحقق من ansatz الأخير.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
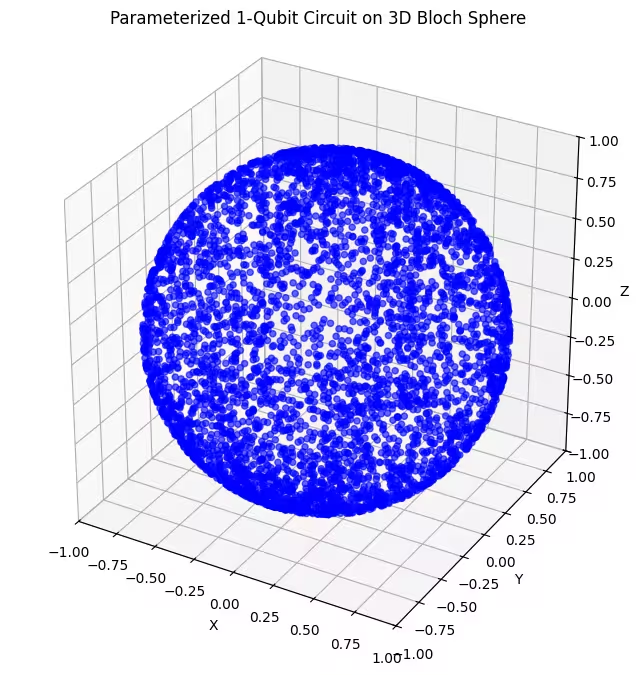
هنا يمكنك رؤية حالات كمومية أكثر انتظاماً في توزيعها، ولّدها ansatz الأخير.
كما ذُكر، أفضل ما يمكنك فعله هو اكتساب المعرفة بالحالة الأساسية التي تسعى إلى إيجادها، واختيار ansatz مناسب لاستكشاف الحالات القريبة منها. على سبيل المثال، لو علمنا أن حالتنا الأساسية قريبة من أحد القطبين، لاخترنا ansatz 2. للتبسيط، سنكتفي بـ ansatz 3، الذي يستكشف كرة بلوخ بأكملها بشكل منتظم.
الآن بعد أن اخترنا الـ ansatz، لنرسم الدائرة.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

الخطوة 2: التحسين للأجهزة المستهدفة
عند تشغيل حساب على حاسوب كمومي حقيقي، لا نهتم فقط بمنطق الدائرة الكمومية. نهتم أيضًا بأمور مثل العمليات التي يمكن لذلك الحاسوب الكمومي بالذات تنفيذها، وأين تقع الكيوبتات التي نستخدمها على الحاسوب الكمومي. هل هي متجاورة؟ هل هي متباعدة؟ لذلك، الخطوة التالية هي إعادة كتابة دائرتنا باستخدام بوابات مناسبة للحاسوب الكمومي الذي سنستخدمه، مع مراعاة تخطيط الكيوبتات. يمكن تحقيق ذلك عبر transpilation - بعد هذه العملية، يمكنك رؤية الـ ansatz البسيط الخاص بنا وقد تحوّل إلى مجموعة مختلفة من البوابات، وسيتم تعيين كيوبتاتنا المجردة إلى كيوبتات فيزيائية على حاسوب كمومي حقيقي.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

يمكنك أن ترى أن بوابات rx, rz في الـ ansatz الخاص بنا تحوّلت إلى سلسلة من بوابات rz, sx، وهي البوابات الأصيلة للـ backend لدينا. كذلك، يمكنك أن ترى أن q0 الخاص بنا تم تعيينه الآن إلى الكيوبت الفيزيائي الخامس. نحتاج أيضًا إلى تعيين هاملتونياننا وفقًا لهذه التغييرات، كما في الكود التالي:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
الخطوة 3: التنفيذ على الأجهزة المستهدفة
حان الوقت الآن لتشغيل VQE على معالج الكم (QPU) الحقيقي. لهذا، نحتاج أولًا إلى دالة تكلفة لعملية التحسين، تحسب قيمة التوقع للهاملتوني مع الحالة الكمومية التي يولّدها الـ ansatz. لا تقلق! لست بحاجة إلى برمجة كل شيء بنفسك. لقد أعددنا دالة لهذا الغرض، وكل ما عليك فعله هو تشغيل الخلية أدناه.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
أخيرًا، نُعدّ المعاملات الأولية للـ ansatz وعملية التحسين الخاصة به. يمكنك ببساطة استخدام أصفار أو قيم عشوائية. لقد اخترنا معاملات أولية أدناه، لكن لا تتردد في التعليق على السطور أو إلغاء تعليقها في الخلية لأخذ عينات عشوائية من المعاملات، موزّعة بشكل منتظم من 0 إلى .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
تهانيّ! لقد أتممت للتو أول تجربة كيمياء كمومية لك بنجاح. يمكننا ملاحظة فارق بين طاقة الحالة الأساسية الدقيقة للهاملتوني وطاقتنا المحسوبة، لكن لأننا استخدمنا تقنية تخفيف أخطاء افتراضية (تصحّح أخطاء القراءة)، فإن الفارق طفيف. هذه بداية ممتازة جدًا!
ملاحظة: يمكنك الحصول على نتيجة أفضل عن طريق تحديد مستوى تخفيف الأخطاء باستخدام resilience_level. القيمة الافتراضية هي 1، وإذا ضبطت قيمة أعلى، فسيستغرق وقتًا أطول من QPU لكنه قد يعيد نتيجة أفضل.
الخطوة 4: ما بعد المعالجة
حان الوقت للنظر في كيفية عمل المُحسِّن الكلاسيكي. شغّل الخلية أدناه وشاهد نمط التقارب.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

بدأنا بقيمة أولية جيدة إلى حد ما، مما أتاح لنا الحصول على قيمة نهائية جيدة في 10 خطوات فقط. يمكنك رؤية ذروات كبيرة وصغيرة، وهذه السمة النموذجية للمُحسِّن COBYLA - يبحث في الفضاء كأنه لا يرى المشهد ويضبط أحجام الخطوات مع كل قياس.
تحقق من فهمك
ما هو ملاحظتك؟ أي جزء من العملية السابقة قابل للتحسين للحصول على نتائج أقرب إلى القيم النظرية، أو أقرب إلى طاقة الحالة الأساسية الدقيقة للهاملتوني؟ ما هي بعض الأمور التي يجب أخذها بعين الاعتبار؟
الإجابة
أول شيء يجب مراعاته هو تغيير مجموعة الأسس المستخدمة في حساب هاملتوني الجزيئات. كما ذُكر سابقًا، طاقة الحالة الأساسية لذرة الهيدروجين هي -0.5 هارتري وهو أمر معروف جيدًا، والأساس STO-6G الذي اخترناه لا يكفي للاشتقاق الدقيق لهذه القيمة.
إن اختيار نوع أساس أكثر تعقيدًا يزيد من عدد الكيوبتات التي يستخدمها الهاملتوني؛ لذلك نحتاج إلى اختيار ansatz أكثر تعقيدًا وملاءمة لمسائل الكيمياء.
الأمر التالي الذي يجب تحسينه هو إدارة الضوضاء في QPU. تقنيات تخفيف الأخطاء الأكثر تقدمًا تعطي نتائج أفضل لكنها قد تستغرق وقتًا أطول. كذلك، فكر في كيفية تأثير shot_number على النتائج.
أخيرًا، يمكن تحقيق أداء تقارب أفضل أيضًا عن طريق تجربة مُحسِّنات مختلفة.
حساب طاقة الحالة الأساسية لجزيء الهيدروجين باستخدام VQE
الآن بعد أن نظرنا في العملية الكلية لـ VQE باستخدام ذرات ، سنحسب طاقة الحالة الأساسية لجزيء بشكل أسرع.
الخطوة 1: تحويل المسألة إلى دوائر كمومية ومؤثرات
هنا نوفر لك أيضًا هاملتونيًا بكيوبت واحد يستخدم أساس STO-6G وتحويل Jordan-Wigner، مع تقليل الكيوبتات باستخدام تماثل الهاملتوني. لاحظ أننا استخدمنا مسافة ذرية بين ذرتَي الهيدروجين تساوي 0.735 .
على خلاف حساب ذرة الهيدروجين المفردة ()، لحساب الحالة الأساسية لجزيء الهيدروجين ()، يجب علينا أيضًا مراعاة قوة التنافر المؤثرة بين نواتَي ذرتَي الهيدروجين، بالإضافة إلى الطاقة المرتبطة بالمدارات الإلكترونية. في هذه الخطوة، سنعطي هذه القيمة كثابت، وسنحسب هذه القيمة فعليًا في مسألة التحقق.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
الخطوة 2: التحسين للأجهزة المستهدفة
بما أن عدد الكيوبتات المستخدم في VQE والهاملتوني السابقَين مطابق لعدد كيوبتات الـ backend المراد استخدامه للتنفيذ، سنستخدم الـ ansatz الموجود وصورته المُحسَّنة.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
الخطوة 3: التنفيذ على الأجهزة المستهدفة
حان الوقت الآن لإجراء الحسابات على QPU الفعلي. كل شيء تقريبًا متطابق، لكننا سنستخدم نقطة بداية مناسبة تتناسب مع الهاملتوني. كذلك، في الجزء التكراري، ستُضبط بعض إعدادات Estimator المستخدم لحساب توقعات الهاملتوني للـ ansatz على QPU بشكل مختلف قليلًا عن الحسابات السابقة. سنناقش هذا التغيير بمزيد من التفصيل في سؤال التحقق.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
على الرغم من أن VQE يوفر نظريًا حدًا أعلى لطاقة الحالة الأساسية الحقيقية، إلا أن التطبيقات العملية على أجهزة الكم الحقيقية أو المحاكاة المضوضاة، إضافة إلى التقريبات المُجراة في إعداد الهاملتوني (كمجموعات الأسس أو تقليل الكيوبتات)، يمكن أن تُدخل أخطاءً تؤدي أحيانًا إلى طاقة قياس أدنى قليلًا من القيمة النظرية الدقيقة أو من مرجع عددي محدد. على الرغم من وجود بعض الأخطاء، تبدو النتائج مرضية، ولا سيما مع العدد الصغير من الخطوات. لنُنهِ الآن حساب VQE هذا بالنظر في كيفية عمل المُحسِّن.
الخطوة 4: ما بعد المعالجة
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

تحقق من فهمك
لنحسب طاقة التنافر النووي لجزيء ، التي أدرجناها كقيمة ثابتة (0.71997 هارتري).

يُرجى استخدام قانون كولوم والوحدة الذرية للتأكد من الحصول على القيمة بوحدة Hartree.
الإجابة
بما أن نواتَي الهيدروجين مشحونتان بشحنة موجبة، فإنهما تتنافران بسبب القوة الكهروستاتيكية. يُوصف هذا التنافر بقانون كولوم:
حيث هي شحنة البروتون، و هي نفاذية الفراغ، و هي المسافة بين النواتين، مقاسةً بالمتر أو بأنصاف قطر بور بوحدة الجول (J).
لحساب هذه الطاقة بالهارتري، نحتاج إلى تحويل المعادلة أعلاه إلى نظام الوحدات الذرية (AU). في AU، و ونصف قطر بور () يساوي 1 ويصبح مقياس الطول الأساسي في AU. مع هذه التبسيطات، يُختزل قانون كولوم إلى:
حيث يجب قياس بأنصاف قطر بور ().
لتحويل الفصل النووي المعطى من إلى ، نحتاج إلى علاقة التحويل هذه:
وبذلك يصبح مساويًا .
لذلك، طاقة التنافر النووي لـ المعطى هي
حساب طاقة التفاعل لـ
الآن دعنا نستخدم ما حصلنا عليه! لقد استخدمت VQE، محلل القيم الذاتية الكمومي التباديلي، لحساب طاقة الحالة الأساسية لذرة ولجزيء . ما تبقى هو استخدام القيم المحسوبة للحصول على طاقة التفاعل لعملية .
طاقة التفاعل هي تغيّر الطاقة الذي يحدث عندما تتفاعل المواد لتكوّن مواد جديدة. تخيّل أنك تبني شيئاً ما: أحياناً تحتاج إلى إدخال طاقة فيه (مثل تكديس المكعبات)، وأحياناً تُطلق الطاقة (مثل كرة تتدحرج من منحدر). في الكيمياء، التفاعلات إما تمتص الطاقة (ماصة للحرارة) أو تطلق الطاقة (طاردة للحرارة).
يمكن حساب طاقة التفاعل لعملية بالصيغة التالية:
بتشغيل الخلية أدناه، دعنا نرى هذا بشكل مرئي. سنستخدم هنا قيمة طاقة الحالة الأساسية الدقيقة لكل هاملتوني، وسنقارن طاقة التفاعل للحل الدقيق ونتائج VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

كما هو موضح في الشكل، على الرغم من وجود بعض الأخطاء، فإن طاقة الحالة الأساسية الدقيقة للهاملتونيين وطاقة التفاعل المحسوبة باستخدام نتائج VQE متشابهة، وتقترب من -0.2 هارتري.
تجدر الإشارة هنا إلى أن طاقة التفاعل لهذه العملية لها قيمة سالبة، مما يعني أن الطاقة تُطلق من خلال العملية، وأن الجزيء الناتج له طاقة أقل من ذرتين منفردتين. 6. الخلاصة
دعنا نلخص ما تعلمناه حتى الآن.
نظرنا أولاً في تقنيتين تقريبيتين مهمتين ضروريتين لحل مشاكل الكيمياء الكمومية: المبدأ التباديلي واختيارات مجموعة الأساس، وكلاهما أساسي لـ VQE. استكشفنا المبدأ التباديلي يدوياً، وحسبنا طاقة الحالة الأساسية للمذبذب التوافقي البسيط.
بعد ذلك، استكشفنا VQE، وهي خوارزمية واسعة الاستخدام لحساب طاقة الحالة الأساسية لنظام كمومي. شغّلنا كوداً لحساب طاقات الحالة الأساسية للهيدروجين الذري () وجزيء الهيدروجين (). على وجه الخصوص، تعلمنا أنه من الضروري الحصول على الهاملتوني الجزيئي المناسب للنظام وتحويله إلى شكل قابل للتنفيذ على الحاسوب الكمومي. رأينا أيضاً أن الـ ansatz، وهو دائرة كمومية مُعلمَّة، ضروري لإعداد حالات كمومية تجريبية داخل VQE، وناقشنا أهمية اختيار بنية دائرة ansatz مناسبة. تعلمنا أيضاً أن VQE تعتمد على عملية تحسين تكرارية باستخدام حاسوب كلاسيكي، توجه الدائرة الكمومية للعثور على حالة الطاقة الأدنى، ورأينا كيف تتقارب العملية.
أخيراً، استخدمنا طاقات الحالة الأساسية المحسوبة لـ و التي تم الحصول عليها من خلال VQE لحساب طاقة التفاعل لعملية .
VQE هي خوارزمية كمومية قوية على المدى القريب، لكن من المهم إدراك قيودها. يعتمد أداء VQE بشكل كبير على اختيار الـ ansatz - إذ يصبح إيجاد ansatz قابل للإعداد بكفاءة يمكنه تمثيل الحالة الأساسية الحقيقية بدقة أمراً صعباً للجزيئات الأكبر والأكثر تعقيداً. علاوة على ذلك، يكون الجهاز الكمومي الحالي عرضة للضوضاء، مما قد يؤثر على دقة نتائج VQE، لا سيما للدوائر الأعمق أو الأعداد الأكبر من الكيوبتات. على الرغم من هذه التحديات، تعمل VQE كخوارزمية تأسيسية، ويستكشف البحث الجاري طرقاً تباديلية أكثر تطوراً وتقنيات تخفيف الأخطاء لتوسيع حدود ما هو ممكن في الكيمياء الكمومية على الحواسيب الكمومية القريبة المدى. على سبيل المثال، يجري تطوير خوارزميات مثل القطرنة الكمومية المعتمدة على العينات (SQD)، التي تستفيد من العينات المأخوذة من الدوائر الكمومية مقترنةً بالقطرنة الكلاسيكية في فضاء جزئي لتحسين تقدير الطاقة ومعالجة بعض القيود التي تواجهها VQE، لا سيما فيما يتعلق بكفاءة القياس ومتانة الضوضاء.
مراجعة وأسئلة
المفاهيم الأساسية:
- الخوارزمية الكمومية التباديلية هي نموذج حوسبة يعمل فيه الحاسوب الكلاسيكي والحاسوب الكمومي معاً لحل مشكلة.
- في VQE، نبدأ بهاملتوني نظامنا ونقوم بتخطيطه على الكيوبتات للتنفيذ على الحاسوب الكمومي. نختار دائرة كمومية مُعلمَّة، وهي الـ ansatz، ونجري قياسات متكررة، مع تغيير معاملات الـ ansatz، حتى نصل إلى أدنى قيمة للطاقة. يتم البحث في فضاء المعاملات باستخدام محسِّن كلاسيكي. لتحقيق نتائج جيدة، من الضروري اختيار ansatz جيد ومحسِّن مناسب.
- طاقة التفاعل هي إجمالي تغيّر الطاقة في التفاعل الكيميائي، تُحدَّد بالفرق بين طاقة المتفاعلات والنواتج.
صح/خطأ
- ينص المبدأ التباديلي على أن القيمة المتوقعة للطاقة لأي دالة موجية تجريبية تكون دائماً أكبر من أو تساوي طاقة الحالة الأساسية الحقيقية.
- مجموعة الأساس هي مجموعة من الدوال تُستخدم لتقريب الدوال الموجية الكمومية.
- VQE هي خوارزمية كمومية تُستخدم لحل معادلة شرودنغر بشكل دقيق لهاملتوني معين.
- في VQE، تُستخدم دائرة كمومية مُعلمَّة (أنساتز) لإعداد دوال موجية تجريبية.
- اختيار المحسِّن في VQE (مثلاً، COBYLA أو SPSA أو ADAM) لا يؤثر على جودة النتيجة.
Estimatorمن Qiskit يُستخدم لحساب القيم المتوقعة للهاملتونيين في VQE مباشرةً.
أسئلة الاختيار من متعدد:
- ما هو الغرض من الهاملتوني في VQE؟
- أ) لتوليد حالات كمومية عشوائية
- ب) لتحديد طاقة الحالات الكمومية
- ج) لتحسين الدوائر الكمومية
- د) لإنشاء التشابك
- ما هو الهدف الرئيسي لخوارزمية VQE؟
- أ) إيجاد طاقة الحالة الأساسية لهاملتوني
- ب) إنشاء التشابك بين الكيوبتات
- ج) تنفيذ بحث غروفر
- د) كسر تشفير RSA
- كم عدد الحالات الكمومية التي تم توليدها في هذا الدفتر لمقارنة الـ ansatz؟
- أ) 100
- ب) 1000
- ج) 5000
- د) 10,000
- لماذا يُعدّ المحسِّن الكلاسيكي ضرورياً في VQE؟
- أ) لإجراء القياسات الكمومية
- ب) تحديث معاملات الـ ansatz لتقليل الطاقة
- ج) لتشابك الكيوبتات
- د) لتوليد العشوائية الكمومية
- لماذا صُمِّم الـ ansatz ليكون مُعلمَّاً؟
- أ) للسماح بإعداد الحالة الكمومية
- ب) للسماح بالبحث في فضاء واسع من الحالات الكمومية
- ج) لتقليل تعقيد الدائرة
- د) لقياس القيم الذاتية مباشرةً
- أي من العبارات التالية هي الأكثر صحة حول اختيار ansatz جيد؟
- أ) يجب أن ينتج الـ ansatz حالات موزعة بالتساوي على كرة بلوخ، وإلا سيفشل.
- ب) يجب تصميم الـ ansatz بما يتناسب مع نظامك لضمان قدرته على توليد حالات قريبة من الحالة الأساسية.
- ج) يجب أن ينتج الـ ansatz حالات عشوائية باستخدام معاملاته التباديلية.
- د) الـ ansatz الأفضل يمتلك دائماً معاملات تباديلية أكثر.
(اختياري) ملحق: عبء المحسِّن حسب تعقيد الـ ansatz
تواجه VQE عدة تحديات معروفة[ref 6]، والتالية مرتبطة بما تعلمناه أعلاه.
- تحديات اختيار الـ ansatz
هناك تحدٍّ متأصل في اختيار الـ ansatz التباديلي الصحيح. توفر ansätze مستوحاة من الكيمياء (مثل UCCSD) دقة فيزيائية لكنها تتطلب دوائر عميقة، في حين أن ansätze الكفاءة الحديثة للعتاد لها دوائر أقل عمقاً لكنها قد تفتقر إلى قابلية التفسير الفيزيائي. كذلك تُدخل كثيرٌ من الـ ansätze معاملاتٍ تباديليةً زائدة لا تسهم كثيراً في تحسين الدقة لكنها تزيد من صعوبة التحسين بشكل ملحوظ.
- صعوبات التحسين
يمكن أن تحتوي مشهد التحسين في VQE على مناطق تتلاشى فيها التدرجات بشكل أسي (الهضاب القاحلة)، مما يجعل من الصعب على المحسِّنات الكلاسيكية تحديث المعاملات التباديلية بكفاءة. لهذا، حاول الباحثون استخدام أنواع مختلفة من المحسِّنات - معتمدة على التدرج وغير معتمدة على التدرج، لكن كلاهما يواجه تحديات. تعاني المحسِّنات المعتمدة على التدرج من الهضاب القاحلة، في حين تتطلب الطرق غير المعتمدة على التدرج عدداً كبيراً من تقييمات الدوال.
- عبء المحسِّن
تحدٍّ معروف آخر هو عبء المحسِّن، المرتبط بحجم المشكلة. تنمو الدوائر الكمومية المطلوبة لـ VQE في العمق والتعقيد مع زيادة حجم المشكلة؛ وهذا عادةً ما يزيد أيضاً من عدد المعاملات المطلوب تحسينها. تصبح عملية التحسين غير قابلة للحساب مع زيادة عدد المعاملات، مما يؤدي إلى تقارب بطيء وصعوبات في إيجاد الحل الأمثل.
هنا سنلقي نظرة على هذه التحديات باستخدام VQE لجزيء ، مع نوعين مختلفين من الـ ansätze.
(ملاحظة: قد يستغرق هذا وقت QPU أطول، لذا لا تتردد في استخدام محاكٍ إن لم يكن لديك وقت كافٍ.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
الآن دعنا نشغل VQE بنقطة بداية مكونة من الواحد فقط، بحد أقصى 20 خطوة، ونقارن تقارب كلا التشغيلين.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

يوضح الرسم البياني أعلاه بجلاء أن عملية التحسين للـ ansatz ذي المتغيرات الأكثر تستغرق وقتاً أطول للوصول إلى تقارب مستقر.
بدلاً من الاعتماد على دوائر كيوبت واحد بسيطة وansatz مباشر، يزداد تعقيد التحسين عند الحاجة إلى دوائر كمومية أكبر وansätze ذات بنية أكثر تعقيداً. يبرز هذا تحدياً معروفاً في VQEs: عبء المحسِّن.
يواصل الباحثون تطوير منهجيات متقدمة متنوعة يمكنها استخدام الحواسيب الكمومية لمشاكل الكيمياء. يمكنك الوصول إلى مجموعة متنوعة من المواد التعليمية في IBM Quantum Learning.
المراجع
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)