تحسين تقدير الطاقة لهاملتوني الكيمياء باستخدام SQD
في هذا البرنامج التعليمي، نُنفّذ نمط Qiskit الذي يوضح كيفية المعالجة اللاحقة للعينات الكمية المشوشة للوصول إلى تقريب للحالة الأساسية لهاملتوني كيمياء: جزيء عند الاتزان في مجموعة الأساس 6-31G. سنتبع نهج القطرنة الكمية القائمة على العينات لمعالجة العينات المأخوذة من نموذج دارة Circuit كمية مؤلفة من 36 qubit (وهي دارة LUCJ في هذه الحالة). ولمراعاة تأثير الضوضاء الكمية، تُستخدم تقنية استرداد التهيئة.
يمكن وصف النمط في أربع خطوات:
- الخطوة 1: التعيين إلى مسألة كمية
- توليد نموذج تقريبي (ansatz) لتقدير الحالة الأساسية
- الخطوة 2: تحسين المسألة
- إجراء عملية Transpile للنموذج التقريبي لتلاؤمه مع الـ Backend
- الخطوة 3: تنفيذ التجارب
- استخلاص عينات من النموذج التقريبي باستخدام primitive الـ
Sampler
- استخلاص عينات من النموذج التقريبي باستخدام primitive الـ
- الخطوة 4: المعالجة اللاحقة للنتائج
- حلقة استرداد التهيئة ذاتية الاتساق
- إجراء معالجة لاحقة لكامل مجموعة عينات السلاسل البتية، مستعينًا بالمعرفة المسبقة بعدد الجسيمات ومتوسط إشغال المدارات المحسوب في آخر تكرار.
- إنشاء دُفعات من العينات الفرعية بصورة احتمالية من السلاسل البتية المستردة.
- إسقاط هاملتوني الجزيء وتطبيق القطرنة عليه في كل فضاء فرعي مُعاين.
- حفظ الحد الأدنى لطاقة الحالة الأساسية الموجودة عبر جميع الدُفعات وتحديث متوسط إشغال المدارات.
- حلقة استرداد التهيئة ذاتية الاتساق
في هذا المثال، يأخذ هاملتوني الإلكترونات المتفاعلة الشكل العام:
/ هما عاملا الإنشاء/الإفناء الفيرميونية المرتبطان بالعنصر من مجموعة الأساس والسبين . أما و فهما التكاملات الإلكترونية أحادية وثنائية الجسيم.
يظهر سير عمل SQD مع استرداد التهيئة ذاتي الاتساق في الرسم التخطيطي التالي.
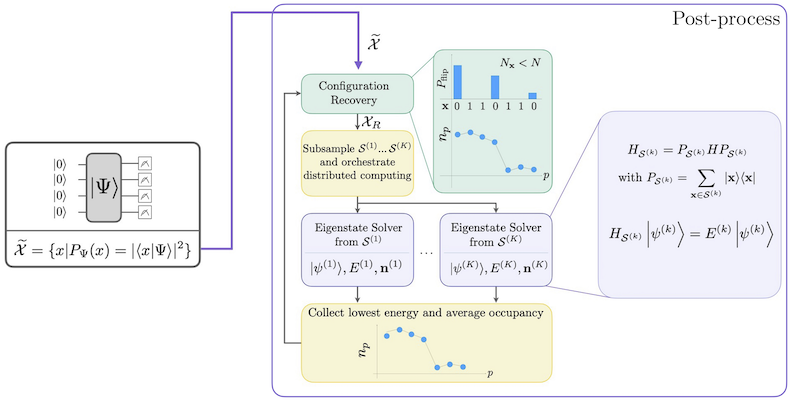
يُعرف SQD بأدائه الجيد حين تكون الحالة الذاتية المستهدفة متفرقة: أي حين تكون دالة الموجة محمولة على مجموعة من حالات الأساس لا يتزايد حجمها أُسّيًا مع حجم المسألة. في هذا السيناريو، تُعطي قطرنة الهاملتوني المُسقَط على الفضاء الفرعي المعرَّف بـ :
تقريبًا جيدًا للحالة الذاتية المستهدفة. يتمثل دور الجهاز الكمي في إنتاج عينات لأعضاء فحسب. أولًا، تُحضّر دارة Circuit كمية الحالة في الجهاز الكمي. يُستخدم ترميز Jordan-Wigner. وبناءً عليه، تُمثّل عناصر الأساس الحسابي حالات Fock (التهيئات/المحددات الإلكترونية). تُعاين الدارة في الأساس الحسابي مما يُنتج مجموعة التهيئات المشوشة . تُمثَّل التهيئات بسلاسل بتية. تُمرَّر المجموعة بعد ذلك إلى كتلة المعالجة الكلاسيكية اللاحقة، حيث تُطبَّق تقنية استرداد التهيئة ذاتية الاتساق. في إطار SQD، يكون دور الجهاز الكمي إنتاج توزيع احتمالي.
الخطوة 1: تعيين المسألة إلى دارة كمية
في هذا البرنامج التعليمي، سنقرّب طاقة الحالة الأساسية لجزيء . سنحدد أولًا الجزيء وخصائصه، ثم ننشئ نموذجًا تقريبيًا LUCJ (local unitary cluster Jastrow) (دارة Circuit كمية) لتوليد عينات من حاسب كمي بغرض تقدير طاقة الحالة الأساسية.
سنحدد أولًا الجزيء وخصائصه.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
بعد ذلك، سننشئ النموذج التقريبي. النموذج LUCJ هو دارة Circuit كمية مُعاملَة، وسنُهيّئه بسعات t2 وt1 المستخلصة من حساب CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
سنستخدم حزمة ffsim لإنشاء النموذج التقريبي وتهيئته بسعات t2 وt1 المحسوبة أعلاه. بما أن جزيئنا يمتلك حالة Hartree-Fock ذات قشرة مغلقة، فسنستخدم المتغير متوازن السبين لنموذج UCJ، وهو UCJOpSpinBalanced.
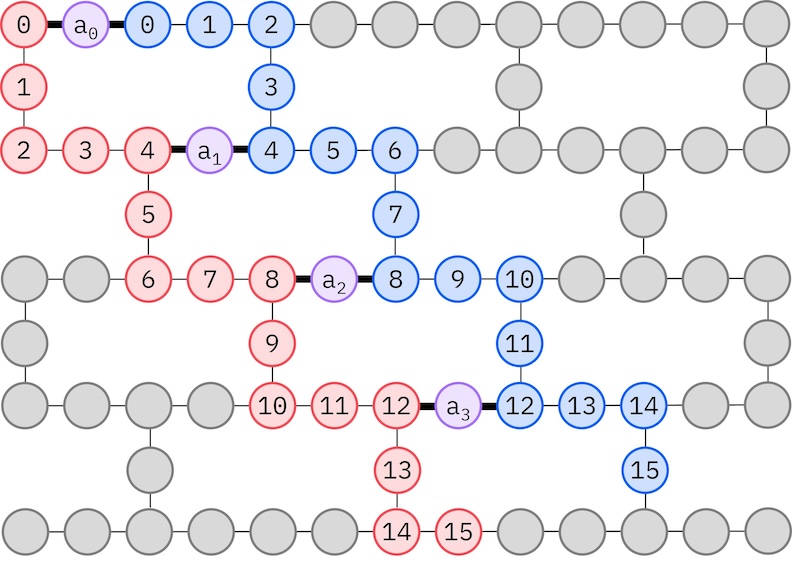
نظرًا لأن العتاد الحديدي المستهدف من IBM يتبع طوبولوجيا السداسي الثقيل (heavy-hex)، سنعتمد نمط zig-zag لتفاعلات qubit. في هذا النمط، ترتبط المدارات (الممثَّلة بـ qubit) التي لها نفس السبين بطوبولوجيا خطية (الدوائر الحمراء والزرقاء) حيث تأخذ كل منها شكل زجزاج بسبب التوصيلية السداسية الثقيلة للعتاد المستهدف. ومرةً أخرى، بسبب طوبولوجيا السداسي الثقيل، تمتلك المدارات ذات السبينات المختلفة توصيلات بين كل مدار رابع (0، 4، 8، إلخ) (الدوائر البنفسجية).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
الخطوة 2: تحسين المسألة
بعد ذلك، سنُحسّن دارتنا لعتاد حديدي مستهدف. نحتاج إلى اختيار جهاز العتاد قبل تحسين دارتنا. سنستخدم Backend وهميًا من 127 qubit من qiskit_ibm_runtime لمحاكاة جهاز حقيقي. للتشغيل على وحدة معالجة كمية فعلية (QPU)، ما عليك سوى استبدال الـ Backend الوهمي بآخر حقيقي. راجع توثيق Qiskit IBM Runtime للمزيد من المعلومات.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
بعد ذلك، نوصي باتباع الخطوات التالية لتحسين النموذج التقريبي وجعله متوافقًا مع العتاد الحديدي.
- اختر qubit فيزيائية (
initial_layout) من العتاد الحديدي المستهدف تتوافق مع نمط الزجزاج الموصوف أعلاه. يؤدي ترتيب qubit بهذا النمط إلى دارة فعّالة ومتوافقة مع العتاد بعدد أقل من البوابات Gates. - أنشئ مدير مسارات مُدرَّج باستخدام الدالة generate_preset_pass_manager من Qiskit مع
backendوinitial_layoutاللذين تختارهما. - اضبط مرحلة
pre_initلمدير المسارات المُدرَّج علىffsim.qiskit.PRE_INIT. يتضمنffsim.qiskit.PRE_INITمسارات Transpiler من Qiskit تُفكّك البوابات Gates إلى دورانات مدارية ثم تدمجها، مما يُنتج عددًا أقل من البوابات Gates في الدارة النهائية. - شغّل مدير المسارات على دارتك.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
الخطوة 3: تنفيذ التجارب
بعد تحسين الدارة لتنفيذها على العتاد الحديدي، نحن مستعدون لتشغيلها على العتاد المستهدف وجمع العينات لتقدير طاقة الحالة الأساسية. بما أنه لدينا دارة واحدة فقط، سنستخدم وضع تنفيذ المهام في Qiskit Runtime وننفّذ دارتنا.
ملاحظة: لقد علّقنا الكود الخاص بتشغيل الدارة على وحدة معالجة كمية (QPU) وأبقيناه للرجوع إليه من قِبَل المستخدم. بدلًا من التشغيل على عتاد حقيقي في هذا الدليل، سنكتفي بتوليد عينات عشوائية مسحوبة من التوزيع المنتظم.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
الخطوة 4: المعالجة اللاحقة للنتائج
الآن، نُشغّل خوارزمية SQD باستخدام الدالة diagonalize_fermionic_hamiltonian. راجع توثيق API للاطلاع على شرح وسيطات هذه الدالة.
يستخدم الحلّال المضمَّن في إضافة SQD تنفيذ PySCF لـ selected CI، وتحديدًا pyscf.fci.selected_ci.kernel_fixed_space. يوضح المثال أدناه أيضًا كيفية تمرير وسيطات الكلمات المفتاحية إلى تلك الدالة عبر الحلّال المضمَّن. هنا نمرر الوسيط max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
الآن، نرسم النتائج.
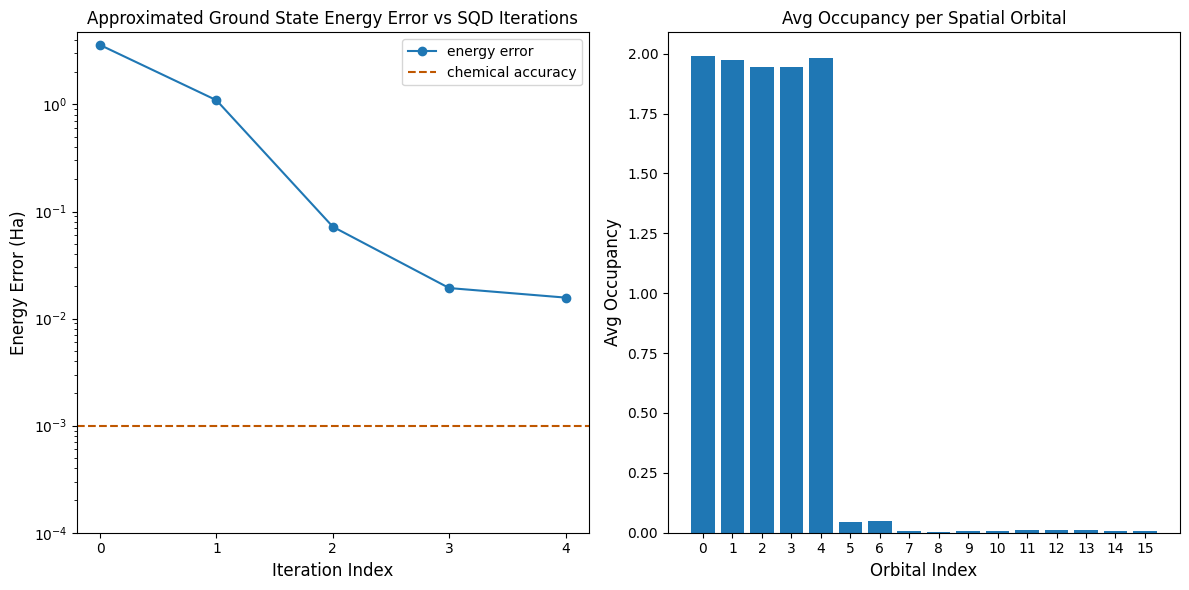
يُظهر الرسم الأول أنه بعد بضعة تكرارات نُقدّر طاقة الحالة الأساسية في حدود ~16 mH (يُقبل عمومًا أن الدقة الكيميائية تبلغ 1 kcal/mol 1.6 mH). تذكّر أن العينات الكمية في هذا العرض التوضيحي كانت ضوضاءً بحتة. تأتي الإشارة هنا من المعرفة المسبقة ببنية الإلكترونات وهاملتوني الجزيء.
يُظهر الرسم الثاني متوسط إشغال كل مدار فضائي بعد التكرار الأخير. يمكننا ملاحظة أن الإلكترونات ذات السبين الصاعد والسبين النازل تشغل المدارات الخمسة الأولى باحتمال مرتفع في حلولنا.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha