القطرنة الكمومية لكريلوف القائم على العينات لنموذج شبكة فيرميوني
تقدير الاستخدام: تسع ثوانٍ على معالج Heron r2 (ملاحظة: هذا تقدير فحسب. قد يختلف وقت التشغيل الفعلي.)
نتائج التعلم
بعد إتمام هذا البرنامج التعليمي، ينبغي على المستخدمين فهم ما يلي:
- كيفية استخدام SQD Qiskit addon لتقريب طاقة الحالة الأساسية لنموذج شبكة باستخدام البتسلاسل المأخوذة من وحدة معالجة كمية (QPU).
- كيفية استخدام ffsim لبناء دوائر التطور الزمني لمحاكاة الفيرميونات.
- كيفية دمج عينات من دوائر متعددة للمعالجة اللاحقة باستخدام خوارزمية القطرنة الكمومية لكريلوف القائم على العينات (SQKD).
المتطلبات الأساسية
نقترح أن يكون المستخدمون على دراية بالمواضيع التالية قبل متابعة هذا البرنامج التعليمي:
- القطرنة الكمومية القائمة على العينات لـ Hamiltonian الكيمياء
- القطرنة الكمومية لكريلوف لـ Hamiltonian الشبكات
- Qiskit primitives
الخلفية النظرية
يوضح هذا البرنامج التعليمي كيفية استخدام القطرنة الكمومية القائم على العينات (SQD) لتقدير طاقة الحالة الأساسية لنموذج شبكة فيرميوني. تحديداً، ندرس نموذج Anderson أحادي الشائبة أحادي البعد (SIAM)، الذي يُستخدم لوصف الشوائب المغناطيسية المضمّنة في المعادن.
يتبع هذا البرنامج التعليمي سير عمل مشابهاً للبرنامج التعليمي المرتبط القطرنة الكمومية القائم على العينات لـ Hamiltonian الكيمياء. غير أن الفارق الجوهري يكمن في طريقة بناء الدوائر الكمية. يستخدم البرنامج التعليمي الآخر نموذجاً تباينياً استدلالياً، وهو جذاب لـ Hamiltonian الكيمياء الذي قد يضم ملايين حدود التفاعل. في المقابل، يستخدم هذا البرنامج التعليمي دوائر تُقرّب التطور الزمني بواسطة الـ Hamiltonian. يمكن أن تكون هذه الدوائر عميقة، مما يجعل هذا النهج أكثر ملاءمةً لتطبيقات نماذج الشبكات. تشكّل متجهات الحالة التي تُعدّها هذه الدوائر أساساً لـ Krylov subspace، وبالتالي تتقارب الخوارزمية بصورة مثبتة وفعّالة نحو الحالة الأساسية في ظل افتراضات مناسبة.
يمكن النظر إلى النهج المستخدم في هذا البرنامج التعليمي باعتباره مزيجاً من التقنيات المستخدمة في SQD وKrylov quantum diagonalization (KQD). يُشار إلى هذا النهج المدمج أحياناً بالقطرنة الكمومية لكريلوف القائم على العينات (SQKD). راجع Krylov quantum diagonalization of lattice Hamiltonians للاطلاع على برنامج تعليمي حول طريقة KQD.
يستند هذا البرنامج التعليمي إلى العمل البحثي "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization"، الذي يمكن الرجوع إليه للاطلاع على مزيد من التفاصيل.
نموذج Anderson أحادي الشائبة (SIAM)
Hamiltonian نموذج SIAM أحادي البعد هو مجموع ثلاثة حدود:
حيث
هنا، هي عوامل الإنشاء/الإفناء الفيرميونية لموقع الحمّام بالسبين ، و هي عوامل الإنشاء/الإفناء لنمط الشائبة، و. أما و و فهي أعداد حقيقية تصف تفاعلات القفز والموقع والتهجين، و عدد حقيقي يحدد الجهد الكيميائي.
لاحظ أن الـ Hamiltonian هو حالة خاصة من Hamiltonian الإلكترون التفاعلي العام،
حيث يتضمن الحدود أحادية الجسيم التربيعية في عوامل الإنشاء والإفناء الفيرميونية، ويتضمن الحدود ثنائية الجسيم الرباعية. بالنسبة لـ SIAM،
ويحتوي على بقية الحدود في الـ Hamiltonian. لتمثيل الـ Hamiltonian برمجياً، نخزّن المصفوفة والموتر .
أساسا الموضع والزخم
نظراً للتماثل الانتقالي التقريبي في ، لا نتوقع أن تكون الحالة الأساسية متفرقة في أساس الموضع (الأساس المداري الذي يُحدَّد فيه الـ Hamiltonian أعلاه). لا يُضمن أداء SQD إلا إذا كانت الحالة الأساسية متفرقة، أي أنها تمتلك وزناً ملحوظاً على عدد صغير فحسب من حالات الأساس الحسابية. لتحسين تفرق الحالة الأساسية، نُجري المحاكاة في الأساس المداري الذي يكون فيه قطرياً. نسمي هذا الأساس أساس الزخم. بما أن هو Hamiltonian فيرميوني تربيعي، يمكن قطرنةه بكفاءة عبر دوران مداري.
التطور الزمني التقريبي بواسطة الـ Hamiltonian
لتقريب التطور الزمني بواسطة الـ Hamiltonian، نستخدم تحليل Trotter-Suzuki من الدرجة الثانية،
في إطار تحويل Jordan-Wigner، يُعادل التطور الزمني بواسطة بوابة CPhase واحدة بين المدارات ذات السبين المتجه لأعلى والسبين المتجه لأسفل عند موقع الشائبة. ولأن هو Hamiltonian فيرميوني تربيعي، يُعادل التطور الزمني بواسطة دوراناً مداريا.
تتشكّل حالات أساس Krylov ، حيث هو بُعد الفضاء الجزئي لـ Krylov، عبر تطبيق متكرر لخطوة Trotter واحدة، وبذلك
في سير عمل SQD التالي، سنأخذ عينات من هذه المجموعة من الدوائر ونعالج المجموعة المجمّعة من البتسلاسل باستخدام SQD. يتناقض هذا النهج مع النهج المستخدم في البرنامج التعليمي المرتبط القطرنة الكمومية القائم على العينات لـ Hamiltonian الكيمياء، حيث كانت العينات تُسحب من دائرة تباينية استدلالية واحدة.
المتطلبات
قبل البدء في هذا البرنامج التعليمي، تأكد من تثبيت ما يلي:
- Qiskit SDK الإصدار 1.0 أو أحدث، مع دعم التمثيل البصري
- Qiskit Runtime الإصدار 0.22 أو أحدث (
pip install qiskit-ibm-runtime) - SQD Qiskit addon الإصدار 0.11 أو أحدث (
pip install qiskit-addon-sqd) - ffsim الإصدار 0.0.72 أو أحدث (
pip install ffsim)
مثال محاكي صغير النطاق
الخطوة 1: تعيين المسألة إلى دائرة كمية
أولاً، نُولّد Hamiltonian نموذج SIAM في أساس الموضع. يُمثَّل الـ Hamiltonian بالمصفوفة والموتر . ثم نُحوّله إلى أساس الزخم. في أساس الموضع، نضع الشائبة في الموقع الأول. غير أننا عند التحويل إلى أساس الزخم، ننقل الشائبة إلى موقع مركزي لتيسير التفاعلات مع المدارات الأخرى.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
بعد ذلك، نُولّد الدوائر لإنتاج حالات أساس Krylov. بالنسبة لكل نوع من أنواع السبين، تُعطى الحالة الابتدائية بوصفها تراكباً لجميع الإثارات الممكنة للإلكترونات الثلاثة الأقرب إلى مستوى Fermi في أقرب 4 أنماط فارغة بدءاً من الحالة ، ويتحقق ذلك بتطبيق سبع بوابات XXPlusYYGate. تُنتج الحالات المتطورة زمنياً عبر تطبيقات متتالية لخطوة Trotter من الدرجة الثانية.
للاطلاع على وصف أكثر تفصيلاً لهذا النموذج وكيفية تصميم الدوائر، يُرجى الرجوع إلى "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
الخطوة 2: تحسين المسألة للتنفيذ الكمي
بعد ذلك، نُحسّن الدائرة لجهاز مستهدف. في الوقت الحالي، سننشئ واجهة خلفية عامة بعدد محدد من الكيوبتات ومجموعة بوابات تتحلل إليها دوائر التطور الزمني بصورة طبيعية.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
الآن، نستخدم Qiskit لتحويل الدوائر إلى الصيغة الملائمة للواجهة الخلفية المستهدفة.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
الخطوة 3: التنفيذ باستخدام Qiskit primitives
بعد تحسين الدوائر للتنفيذ على الأجهزة، أصبحنا مستعدين لتشغيلها على الجهاز المستهدف وجمع العينات لتقدير طاقة الحالة الأساسية. بعد استخدام الأداة الأولية Sampler لأخذ عينات من البتسلاسل في كل دائرة، نجمع جميع النتائج في قاموس عدّ واحد ونرسم رسماً بيانياً لأكثر 20 بتسلسلة عينةً شيوعاً.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
الخطوة 4: المعالجة اللاحقة وإعادة النتيجة إلى الصيغة الكلاسيكية المطلوبة
الآن، نُشغّل خوارزمية SQD باستخدام الدالة diagonalize_fermionic_hamiltonian. راجع توثيق API للاطلاع على شرح وسيطات هذه الدالة.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
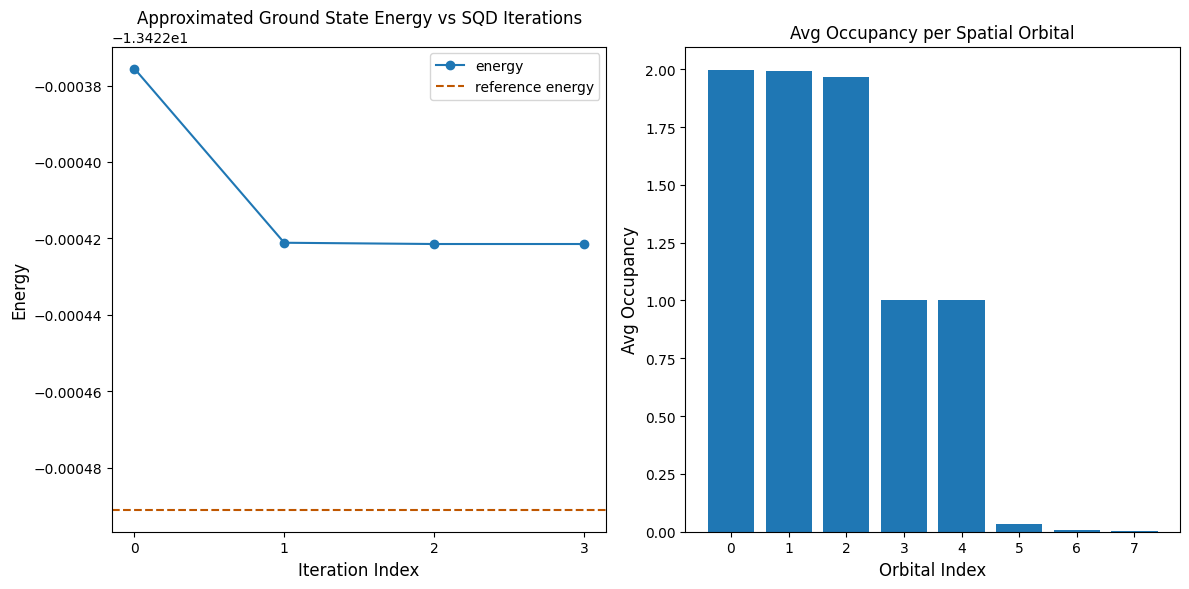
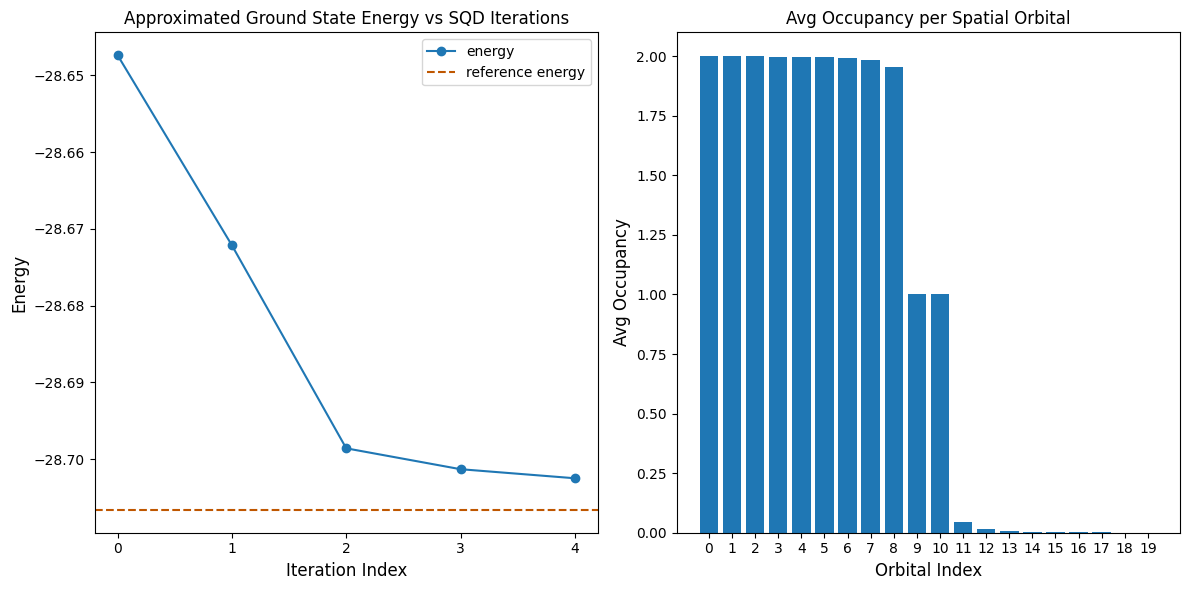
يعرض الكود التالي نتائج في مخططين. يُظهر المخطط الأول الطاقة المحسوبة كدالة لعدد تكرارات استرداد التهيئة، ويُظهر المخطط الثاني متوسط إشغال كل مدار مكاني بعد التكرار الأخير. بما أن هذه مسألة صغيرة جداً، فإن التكرار الأول يُقرّبنا بالفعل من الطاقة الدقيقة (لاحظ مقياس المحور الصادي).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
التحقق من الطاقة
الطاقة التي تُعيدها SQD مضمونة بوصفها حداً أعلى لطاقة الحالة الأساسية الحقيقية. يمكن التحقق من قيمة الطاقة لأن SQD تُعيد أيضاً معاملات متجه الحالة الذي يُقرّب الحالة الأساسية. يمكنك حساب الطاقة من متجه الحالة باستخدام مصفوفتي كثافة الاختزال أحادية وثنائية الجسيم (الاختزال أحادي الجسيم وثنائي الجسيم)، كما هو موضح في كتلة الكود التالية.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
مثال الأجهزة الكبيرة النطاق
الآن، نُشغّل مثالاً أكبر على وحدة معالجة كمية حقيقية (QPU). كطاقة مرجعية، نستخدم نتائج حساب DMRG أُجري بصورة منفصلة.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
الخطوات التالية
إذا أثار هذا العمل اهتمامك، قد يعجبك الاطلاع على المواد التالية:
- القطرنة الكمومية القائمة على العينات لـ Hamiltonian الكيمياء - برنامج تعليمي مرتبط يستخدم نموذجاً تباينياً استدلالياً بدلاً من دوائر Trotter
- القطرنة الكمومية لكريلوف لـ Hamiltonian الشبكات - برنامج تعليمي حول طريقة KQD
- توثيق SQD addon API - مرجع لدالة
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - الورقة البحثية التي يستند إليها هذا البرنامج التعليمي