القطرنة الكمومية القائمة على العينات لهاميلتوني كيمياء
تقدير الاستخدام: أقل من دقيقة واحدة على معالج Heron r2 (ملاحظة: هذا تقدير فقط. قد يختلف وقت تشغيلك الفعلي.)
نتائج التعلم
بعد الانتهاء من هذا البرنامج التعليمي، ينبغي أن يفهم المستخدمون:
- كيفية استخدام إضافة SQD لـ Qiskit لتقريب طاقة الحالة الأرضية لنظام جزيئي باستخدام سلاسل بت مأخوذة من وحدة معالجة كمومية (QPU).
- كيفية استخدام ffsim لبناء دائرة نموذج المجموعة العنقودية الوحدوية المحلية Jastrow (LUCJ) لمحاكاة الكيمياء الكمية.
المتطلبات الأساسية
نوصي المستخدمين بالتعرف على المواضيع التالية قبل البدء بهذا البرنامج التعليمي:
- الكيمياء الكمية والتكميم الثاني
- استخدام الأولي Sampler لأخذ عينات من الدوائر الكمية
الخلفية
في هذا البرنامج التعليمي، نوضح كيفية معالجة العينات الكمية الصاخبة لتقريب الحالة الأرضية لجزيئة النيتروجين عند طول الرابطة في حالة الاتزان، وذلك باستخدام إضافة SQD لـ Qiskit لتنفيذ خوارزمية القطرنة الكمومية القائمة على العينات (SQD). يمكن الاطلاع على مزيد من التفاصيل حول البرنامج في التوثيق المقابل، بما في ذلك مثال بسيط للبدء.
يُوصى بهذا البرنامج التعليمي للمستخدمين المألوفين بالكيمياء الكمية؛ وتحديدًا الإلمام بإيجاد طاقات الحالة الأرضية للجزيئات. للاطلاع على شرح تفصيلي لسير العمل، يمكن الرجوع إلى دورة خوارزمية القطرنة الكمومية.
SQD هي تقنية لإيجاد القيم الذاتية والمتجهات الذاتية للمؤثرات الكمية، مثل هاميلتوني النظام الكمي، باستخدام الحوسبة الكمية والحوسبة الكلاسيكية الموزعة معًا. تُستخدم الحوسبة الكلاسيكية الموزعة لمعالجة العينات المحصّلة من المعالج الكمي، وإسقاط هاميلتوني الهدف وقطرنةه في الفضاء الجزئي الذي تمتد إليه. تتضمن سير عمل SQD الخطوات التالية:
- اختيار نموذج الدائرة (ansatz) وتطبيقه على حاسوب كمي على حالة مرجعية (في هذه الحالة، حالة هارتري-فوك).
- أخذ عينات من سلاسل البت الناتجة عن الحالة الكمية.
- تشغيل إجراء استعادة التكوين ذاتي التناسق على سلاسل البت للحصول على تقريب للحالة الأرضية.
يُعرف بأن SQD يعمل بشكل جيد عندما تكون الحالة الذاتية المستهدفة متفرقة: أي أن دالة الموجة تُدعم في مجموعة من الحالات الأساسية التي لا يتزايد حجمها أسيًا مع حجم المسألة.
الكيمياء الكمية
يمكن كتابة هاميلتوني النظام الجزيئي على النحو التالي:
حيث و هي أعداد مركبة تُسمى التكاملات الجزيئية، ويمكن حسابها من مواصفات الجزيئة باستخدام برنامج حاسوبي. في هذا البرنامج التعليمي، نحسب هذه التكاملات باستخدام حزمة البرامج PySCF.
للاطلاع على تفاصيل حول كيفية اشتقاق الهاميلتوني الجزيئي، يُرجى الرجوع إلى أحد كتب الكيمياء الكمية (على سبيل المثال، Modern Quantum Chemistry بقلم Szabo وOstlund). للحصول على شرح عالي المستوى حول كيفية تعيين مسائل الكيمياء الكمية على الحواسيب الكمية، اطلع على المحاضرة Mapping Problems to Qubits من Qiskit Global Summer School 2024.
نموذج المجموعة العنقودية الوحدوية المحلية Jastrow (LUCJ)
تتطلب SQD نموذج دائرة كمية (ansatz) لأخذ العينات منه. في هذا البرنامج التعليمي، سنستخدم نموذج المجموعة العنقودية الوحدوية المحلية Jastrow (LUCJ) نظرًا لما يجمعه من التحفيز الفيزيائي والملاءمة للأجهزة. وسنستخدم ffsim لبناء دائرة النموذج.
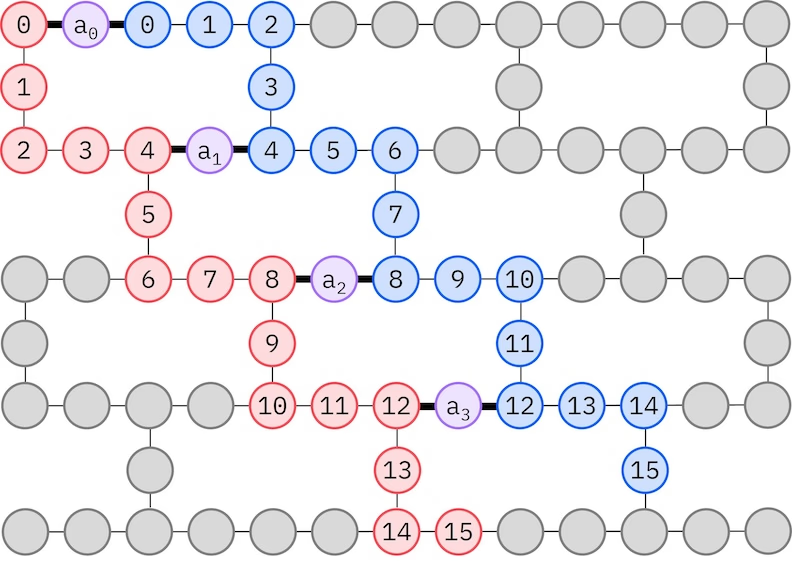
يتكيّف نموذج LUCJ مع وحدات معالجة الكيوبتات ذات الاتصالية المحدودة، إذ تُعيَّن مدارات السبين على كيوبتات بحيث لا يستلزم النموذج توجيهًا ببوابات SWAP. تمتلك أجهزة IBM® طبولوجيا كيوبتات بشبكة سداسية ثقيلة (heavy-hex)، وفي هذه الحالة يمكننا اعتماد نمط "متعرج"، موضح أدناه. في هذا النمط، تُعيَّن المدارات ذات السبين نفسه على كيوبتات بطبولوجيا خطية (الدوائر الحمراء والزرقاء)، وتوجد وصلة بين مدارات ذات سبين مختلف عند كل رابع مدار مكاني، مع تيسير الوصلة بكيوبت إضافي (دوائر بنفسجية).
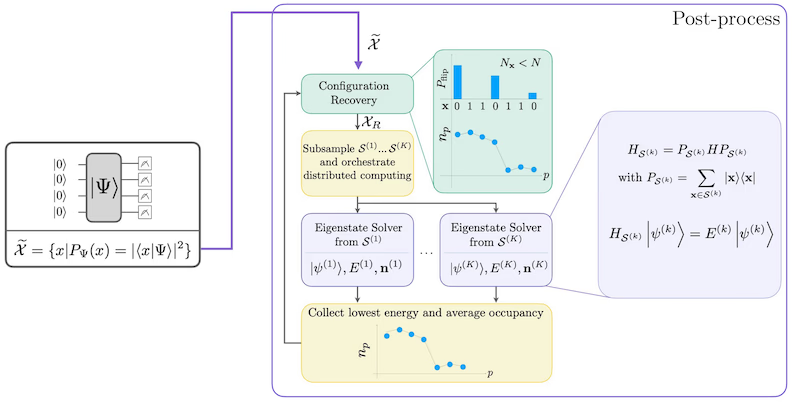
استعادة التكوين ذاتي التناسق
صُمِّم إجراء استعادة التكوين ذاتي التناسق لاستخلاص أكبر قدر ممكن من الإشارة من العينات الكمية الصاخبة. بما أن الهاميلتوني الجزيئي يحفظ عدد الجسيمات والزخم الزاوي الذاتي Z، فمن المنطقي اختيار نموذج دائرة يحفظ هذه التماثلات أيضًا. عند تطبيقه على حالة هارتري-فوك، تمتلك الحالة الناتجة عددًا ثابتًا من الجسيمات وزخمًا زاويًا ذاتيًا Z ثابتًا في الحالة الخالية من الضوضاء. لذلك، يجب أن يكون لنصفَي سبين- وسبين- من أي سلسلة بت مأخوذة من هذه الحالة وزن هامينغ مماثل لما في حالة هارتري-فوك. بسبب وجود الضوضاء في المعالجات الكمية الحالية، ستنتهك بعض سلاسل البت المقيسة هذه الخاصية. أبسط صيغة للاختيار اللاحق هو تجاهل سلاسل البت هذه، لكن ذلك مُبذِّر لأن تلك السلاسل قد لا تزال تحتوي على بعض الإشارة. يحاول إجراء الاستعادة ذاتية التناسق استرداد بعض من تلك الإشارة في المعالجة اللاحقة. الإجراء تكراري ويتطلب كمدخلات تقديرًا لمتوسط إشغالات كل مدار في الحالة الأرضية، والذي يُحسب أولًا من العينات الخام. يُشغَّل الإجراء في حلقة، وتشمل كل تكرارة الخطوات التالية:
- لكل سلسلة بت تنتهك التماثلات المحددة، يتم قلب بتاتها بإجراء احتمالي مصمم لتقريب سلسلة البت من التقدير الحالي لمتوسط إشغالات المدارات، للحصول على سلسلة بت جديدة.
- جمع كل سلاسل البت القديمة والجديدة التي تستوفي التماثلات، وأخذ عينات فرعية من مجموعات بحجم ثابت محدد مسبقًا.
- لكل مجموعة فرعية من سلاسل البت، إسقاط الهاميلتوني في الفضاء الجزئي الممتد بالمتجهات الأساسية المقابلة (انظر القسم السابق للاطلاع على وصف هذه المتجهات الأساسية)، وحساب تقدير الحالة الأرضية للهاميلتوني المُسقَط على حاسوب كلاسيكي.
- تحديث تقدير متوسط إشغالات المدارات بتقدير الحالة الأرضية ذي الطاقة الأدنى.
مخطط سير عمل SQD
يُصوَّر سير عمل SQD في المخطط التالي:
المتطلبات
قبل البدء في هذا البرنامج التعليمي، تأكد من تثبيت ما يلي:
- Qiskit SDK الإصدار 1.0 أو أحدث، مع دعم التصوير البياني
- Qiskit Runtime الإصدار 0.22 أو أحدث (
pip install qiskit-ibm-runtime) - إضافة SQD لـ Qiskit الإصدار 0.11 أو أحدث (
pip install qiskit-addon-sqd) - ffsim الإصدار 0.0.75 أو أحدث (
pip install ffsim)
الإعداد
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
مثال المحاكي على نطاق صغير
في هذا البرنامج التعليمي، سنجد تقريبًا للحالة الأرضية لجزيئة النيتروجين بالقرب من مسافة الرابطة في حالة الاتزان. نستخدم أولًا مجموعة أساسية صغيرة STO-6G كي نتمكن من محاكاة التجربة والتحقق من صحتها.
الخطوة 1: تعيين المدخلات الكلاسيكية إلى مسألة كمية
أولًا، نُحدد الجزيئة وخصائصها.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
قبل بناء دائرة نموذج LUCJ، نجري أولًا حساب CCSD في خلية الكود التالية. ستُستخدَم سعات و من هذا الحساب لتهيئة معاملات النموذج.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
الآن، نستخدم ffsim لإنشاء دائرة النموذج. بما أن جزيئتنا لها حالة هارتري-فوك ذات قشرة مغلقة، نستخدم المتغير المتوازن للسبين من نموذج UCJ، UCJOpSpinBalanced. نضع optimize=True في طريقة from_t_amplitudes لتمكين التحليل العاملي المزدوج "المضغوط" لسعات (انظر نموذج المجموعة العنقودية الوحدوية المحلية Jastrow (LUCJ) في توثيق ffsim للتفاصيل).
بما أن نموذج LUCJ يتكيّف مع الاتصالية المتاحة في وحدة معالجة الكيوبتات، يجب تهيئة الواجهة الخلفية لوحدة معالجة الكيوبتات قبل إنشاء النموذج. سننشئ الآن واجهة خلفية عامة بخريطة اقتران heavy-hex ومجموعة بوابات تتحلل إليها طبيعيًا دائرة نموذج LUCJ. ثم سنستخدم ffsim.qiskit.generate_lucj_pass_manager لإنشاء مدير تمرير متخصص في تحويل نموذج LUCJ إلى الواجهة الخلفية المعطاة وفق تخطيط "zig-zag" الموصوف في قسم الخلفية حول نموذج LUCJ. تستخدم هذه الدالة إرشادًا للتسجيل لتقليل الأخطاء المرتبطة بالتخطيط المختار، وهو أمر مهم عند استخدام وحدة معالجة كيوبتات حقيقية أو محاكٍ بنموذج ضوضاء. إضافةً إلى إعادة مدير التمرير، تُعيد هذه الدالة أيضًا أزواج الاقتران بين مدارات alpha-beta التي يمكن تنفيذها على الجهاز. إذا لم تكن جميع الأزواج قابلة للتنفيذ، تُصدر تحذيرًا.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
الخطوة 2: التحسين للتنفيذ على الأجهزة الكمية
بعد ذلك، نُحسِّن الدائرة للتنفيذ على الجهاز الهدف. عادةً تتضمن هذه الخطوة تهيئة الواجهة الخلفية للجهاز ومدير التمرير المناسب. غير أنه نظرًا لتكيّف نموذج LUCJ مع اتصالية الجهاز، فقد أجرينا هاتين العمليتين في الخطوة السابقة. كل ما يتبقى هو تشغيل مدير التمرير على الدائرة لتحويلها إلى دائرة ISA قابلة للتنفيذ مباشرةً على وحدة معالجة الكيوبتات.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
الخطوة 3: التنفيذ باستخدام الأوليات الكمية في Qiskit
بعد تحسين الدائرة للتنفيذ على الجهاز، أصبحنا جاهزين لتشغيلها على الجهاز الهدف وجمع العينات لتقدير طاقة الحالة الأرضية. بما أن لدينا دائرة واحدة فقط، سنستخدم وضع تنفيذ المهمة في Qiskit Runtime وتنفيذ دائرتنا.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
الخطوة 4: المعالجة اللاحقة وإعادة النتيجة بالصيغة الكلاسيكية المطلوبة
مقياس مفيد للحكم على جودة مخرجات وحدة معالجة الكيوبتات هو عدد التكوينات الصالحة المُعادة. التكوين الصالح هو الذي يمتلك عدد الجسيمات وزخم السبين Z الصحيحَين، أي أن النصف الأيمن من سلسلة البت له وزن هامينغ مساوٍ لعدد الإلكترونات ذات السبين الصاعد، والنصف الأيسر له وزن هامينغ مساوٍ لعدد الإلكترونات ذات السبين الهابط. تحسب الخلية التالية نسبة التكوينات المأخوذة عينةً منها التي تعدّ صالحة.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
جميع سلاسل البت صالحة لأننا نأخذ العينات من المحاكي الخالي من الضوضاء. عند التشغيل على وحدة معالجة كيوبتات حقيقية مع ضوضاء، ستكون النسبة أقل من واحد، لكن نأمل أن تكون أكبر من النسبة المتوقعة لو أُخذت سلاسل البت عشوائيًا بالتساوي، والتي تُحسب في الخلية التالية.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
الآن، نُقدِّر طاقة الحالة الأرضية للهاميلتوني باستخدام دالة diagonalize_fermionic_hamiltonian. تُجري هذه الدالة إجراء استعادة التكوين ذاتي التناسق لتحسين العينات الكمية الصاخبة تكراريًا وتحسين تقدير الطاقة. نمرر دالة رد نداء (callback) كي نتمكن من حفظ النتائج الوسيطة للتحليل لاحقًا. انظر توثيق API للاطلاع على شروح الوسائط الخاصة بـ diagonalize_fermionic_hamiltonian.
نستخدم هنا الوسيط initial_occupancies في diagonalize_fermionic_hamiltonian لتحديد تكوين هارتري-فوك كتخمين أولي لإشغالات المدارات في الحالة الأرضية. هذا النهج معقول للأنظمة التي تمتلك فيها الحالة الأرضية دعمًا كبيرًا على تكوين هارتري-فوك، لكنه قد لا يكون مناسبًا في حالات أخرى، وإن كانت الأساليب الحسابية الأكثر تقدمًا قد تعطي تخمينات أولية أفضل في تلك الحالات. يتيح تحديد initial_occupancies أيضًا تشغيل استعادة التكوين حتى لو لم تُؤخذ أي تكوينات صالحة كعينات، وهو ما قد يحدث عند أخذ العينات من دائرة كبيرة على وحدة معالجة كيوبتات مشوشة. بدون هذا الوسيط، ستفشل استعادة التكوين وتُطلق خطأ إذا لم تُقدَّم أي تكوينات صالحة.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
تصوير النتائج
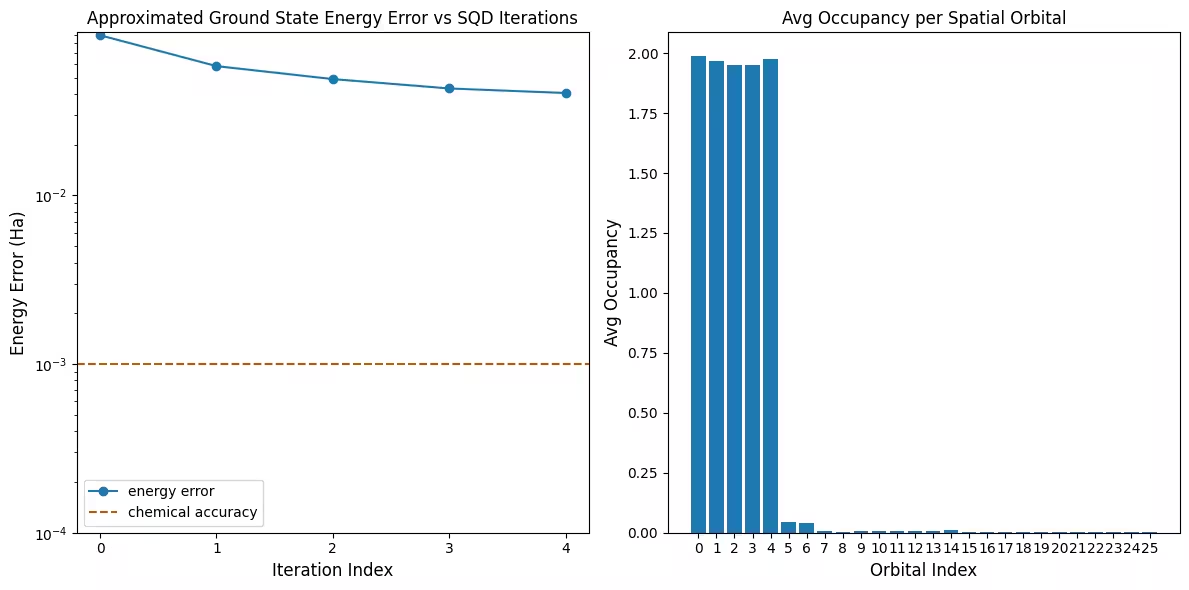
يُظهر الرسم الأول أنه في هذه المحاكاة نكون ضمن 1 mH من الإجابة الدقيقة بعد التكرار الأول (الدقة الكيميائية مقبولة عمومًا بـ 1 kcal/mol 1.6 mH). هذا نظام صغير، ولأن العينات خالية من الضوضاء، لا تكون استعادة التكوين ضرورية. في نظام أكبر يعمل على وحدة معالجة كيوبتات مشوشة، قد تكون تكرارات استعادة التكوين المتعددة ضرورية، وقد تكون الدقة النهائية أسوأ. بصفة عامة، يمكن تحسين الطاقة بالسماح بمزيد من تكرارات استعادة التكوين أو بزيادة عدد العينات في كل دُفعة.
يُظهر الرسم الثاني متوسط إشغال كل مدار مكاني بعد التكرار الأخير. يمكننا أن نرى أن الإلكترونات ذات السبين الصاعد والهابط تشغل المدارات الخمس الأولى باحتمالية عالية في حلولنا.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

مثال الأجهزة الحقيقية على نطاق واسع
الآن، نُشغِّل مثالًا أكبر على أجهزة كمية حقيقية. سنشتق هنا فضاءً نشطًا لجزيئة النيتروجين من المجموعة الأساسية cc-pVDZ.
الخطوات 1-4
هنا نجمع جميع الخطوات في سير عمل واحد على نطاق أوسع، يُشغَّل بعد ذلك على أجهزة كمية حقيقية.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
الخطوات التالية
إذا وجدت هذا العمل مثيرًا للاهتمام، فقد تجد المواد التالية مفيدة:
- القطرنة الكمومية القائمة على عينات Krylov لنموذج شبكة فيرميونية - برنامج تعليمي مرتبط يستخدم دوائر التطور الزمني بدلًا من النموذج التغايري
- توسيع سير عمل كيمياء SQD باستخدام محلل Dice - صفحة توضح كيفية استخدام برنامج Dice الأكثر كفاءة للقطرنة
- توثيق API لإضافة SQD - مرجع دالة
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - الورقة البحثية التي يستند إليها هذا البرنامج التعليمي